
高固含量NTO 的连续流合成及性能表征
2024-03-27 07:39赵登鹏卢欢唱张祯琦黄靖伦田均均荆苏明范桂娟
含能材料 2024年3期
杨 炜,赵登鹏,,卢欢唱,张祯琦,黄靖伦,马 卿,田均均,荆苏明,范桂娟
(1.中国工程物理研究院化工材料研究所, 四川 绵阳 621999; 2.中北大学环境与安全工程学院, 山西 太原 030051)
0 引 言
复杂恶劣环境对弹药安全性提出了更高要求,高能低感炸药对于提升弹药安全性和毁伤威力至关重要[1]。3-硝基-1,2,4-三唑-5-酮(NTO)[2-3]作为一种高能量、不敏感的低成本炸药,其密度达到了1.93 g·cm-3。由于其具有优异的机械感度(撞击感度H50=291 cm)、较高的能量(爆速Vd=8510 m·s-1)和良好的热稳定性(分解温度为275.5 ℃)[4],被美、法等国用于多种不敏感炸药配方研制和生产。法国XF11585 不敏感熔铸炸药已成功装填多种炮弹,并在2016 年新建成一条NTO 连续化生产线,年产能力超过200 吨[5]。NTO 优异的高能量和不敏感特性使得其在不敏感弹药上具有广阔应用前景。然而,NTO 传统釜式硝化工艺[6-7]是以2,4-二氢-1,2,4-三唑-5-酮(TO)为原材料,在98%硝酸条件下合成,该硝化反应体系的绝热温升ΔTad高达531.3 K[8],若反应放热无法有效移除,体系温度持续升高将导致冒料甚至爆炸等风险,因此,有必要开发更加安全、高效的NTO 连续合成技术,提升硝化工艺的本质安全性。
连续流技术利用通道的特殊结构和大比表面积,可以实现反应体系传质传热效率的显著增加,防止热量累积和提升质量稳定性,已经在药物、材料等领域取得了快速的发展[9-11]。炸药合成作为一种高危化学过程,涉及硝化、氧化等危险反应类型,存在放热量大、在线炸药量大等突出安全风险。通过连续流技术解决炸药合成工艺的安全性与效率问题,已成为含能材料领域的研究热点[12]。Zukerman[13-14]等通过微反应器实现了高能低感炸药2,6-二氨基-3,5-二硝基吡嗪-1-氧化物(LLM-105)的连续硝化合成,可以有效控制反应过程放热,避免釜式冒料等风险。Anderson[15]报道了从2,4-二 硝 基 甲 苯(DNT)到2,4,6-三 硝 基 甲 苯(TNT)的自动化克量级连续合成。武碧栋[16]利用微流 控 技 术 合 成 了3,3´-二 氨 基-4,4´-氧 化 偶 氮 呋 咱(DAAF),在室温条件下以89.96%收率得到粒径分布更窄、安全性能更好的DAAF。刘卫孝[17]开展微通道反应器中二缩三乙二醇二硝酸酯(TEGDN)合成工艺研究,产率和选择性得到提高。齐秀芳[18]设计了基于混沌混合式微混合器的微反应系统,实现了硝化甘油的连续合成工艺。朱朋[19]开发了一种新型微反应系统,合成了晶体形态更好、粒度分布更窄的含能金属盐三硝基间苯二酚钡(BaTNR)和三硝基间苯二酚铅(LTNR)。李斌栋[20]利用内交叉多层式微反应器合成了1-甲基-4,5-二硝基咪唑(4,5-MDNI),单位时间及单位体积的制备能力显著提高。
由于连续流合成技术具有出色的传质传热效率和低在线炸药量,可很好地解决含能材料釜式合成工艺的热安全风险,但由于固体产物反应析出时会导致管道内部结垢甚至堵塞,目前含能材料合成以液相反应体系为主。由于NTO 合成过程中生成的大量固体产物极易堵塞微通道,对高固含量反应体系特点的NTO连续流硝化合成研究还较少。为实现NTO 合成工艺的连续化、安全化,本研究针对其不同阶段的反应特点设计了新型连续流反应系统,对合成工艺以及产品纯度、形貌、晶型、热性能、机械感度进行了研究,并与常规方法合成的NTO 性能和工艺进行了对比。
1 实验部分
1.1 试剂与仪器
2,4-二氢-1,2,4-三唑-5-酮,97%,毕得医药;硝酸,98%,科隆试剂;乙腈,色谱纯,诺尔施;去离子水,纯水机自制。
DP-H50 双柱塞高压恒流输液泵,欧世盛(北京)科技有限公司;LAMBDA Doser 固体加料器,德国LAMBDA 公司;管式反应器,自制;循环温度控制器,德国Lauda;Avance 600 核磁共振波谱仪,德国Bruker公司;高效液相色谱仪,美国Agilent 公司;STA449 TG-DSC 热分析仪,德国耐驰公司;D8 Advance 型粉末衍射仪,德国Bruker 公司;Scope A1 光学显微镜,德国ZEISS 公司;傅里叶变换红外吸收光谱仪,美国Perkin Elmer 公司;元素分析仪,德国Elementar 公司;分析天平,瑞士Mettler Toledo 公司。
1.2 连续流反应系统设计
NTO 硝化反应前期硝化体系为液体均相,随着反应转化率逐渐增加并达到饱和浓度,反应后期会析出大量固体并伴随放热和发泡现象。大量固体析出会造成管道堵塞,无法实现微通道连续制备,同时,气泡还将导致反应器有效持液量下降,缩短硝化反应有效停留时间。针对NTO 硝化过程的体系固含量特点,本研究设计并搭建了NTO 连续流反应系统,以实现不同阶段的连续硝化合成。装置包括了自动配料单元、硝化反应单元、过滤单元和控制系统4 个部分(如图1 所示):配料单元由固体自动加料机、酸液高压输液泵和溶解釜组成,硝化反应单元包括反应液高压输液泵、温度控制器、盘管反应器、管式反应器构成,过滤单元包括过滤瓶、真空装置,控制系统通过柔性工艺编辑软件统一控制加料机、输液泵、温控设备参数。其中,盘管反应器由PFA 管(2 mm×3 mm)组成,管式反应器(6 mm×8 mm)内部包含机械搅拌元件以实现固料的高效混合和输送,防止固体堵塞管路。
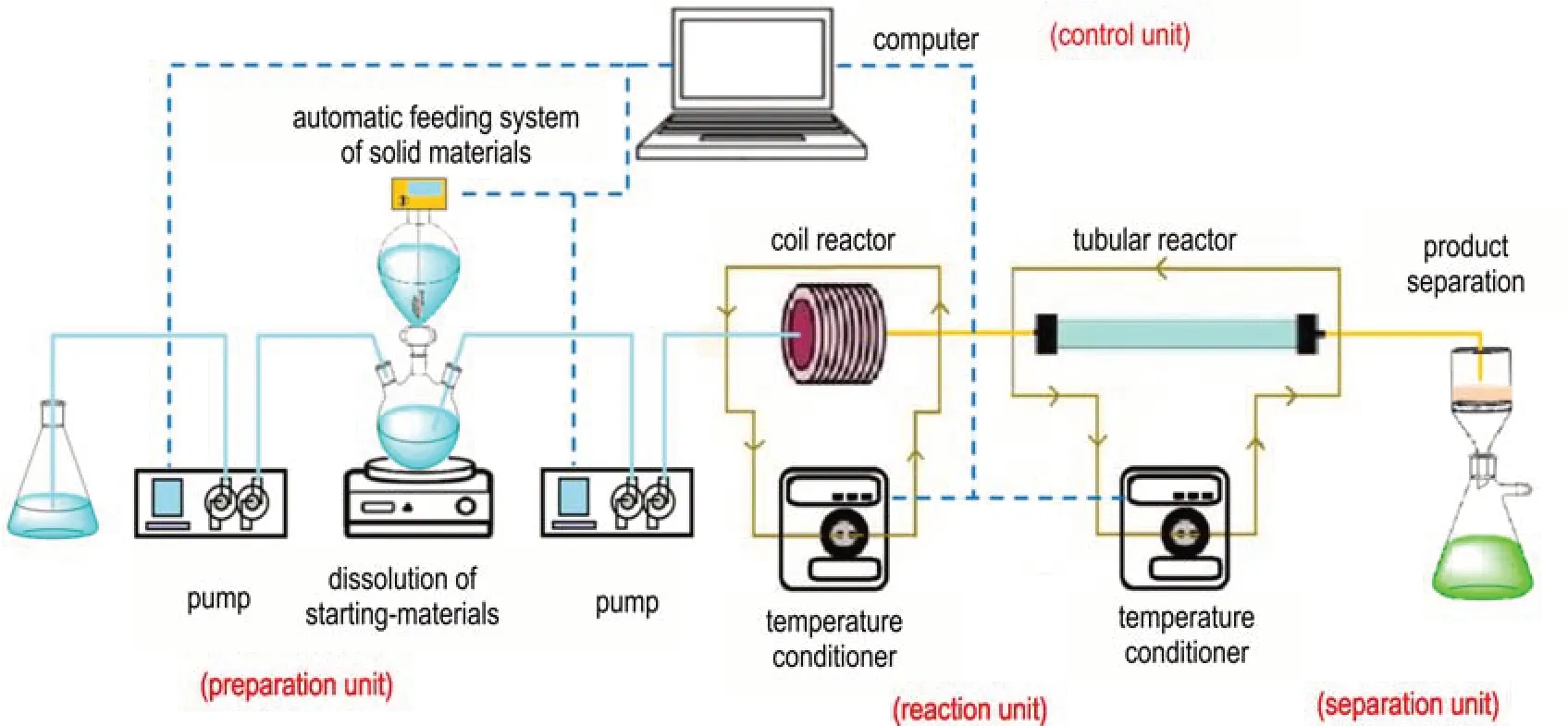
图1 NTO 的连续流反应系统装置图Fig.1 Schematic diagram of the continuous flow synthesis system for NTO
1.3 实验步骤
连续流合成NTO:称取200 g 98%硝酸加入锥形烧瓶中,并在冰水浴条件下缓慢加入30.6 g 纯水,搅拌均匀得到85%硝酸溶液。将100 g TO 固体粉末加入固体加料器中。开启温度控制器,设定工艺温度后启动循环控温。启动溶解釜磁力搅拌器,并进行冰水浴控温。开启酸液高压输液泵将85%硝酸溶液泵入溶解釜,开启固体加料机连续加入TO 固体粉末至溶解釜,设定85%硝酸溶液的流速为13.4 mL·min-1和TO加料速度约为4.0 g·min-1。开启反应液高压输液泵将反应液依次泵入盘管反应器和管式反应器,设定反应液流速为14.4 mL·min-1。反应结束后,反应液直接流入抽滤瓶中过滤。滤饼经冰水洗涤后,得到白色NTO 固体粉末。
釜式合成NTO:参考文献[3],冰浴条件下向500 mL三口烧瓶中加入发烟硝酸222 mL,搅拌10 min 使反应液降温至0 ℃,少量多次加入89 g TO 固体,控制加料温度不超过10 ℃,加完后升温至室温继续反应1.5 h。反应结束后,将反应液倒入抽滤瓶中过滤。滤饼经冰水洗涤后,得到白色NTO 固体粉末。
1.4 表征与测试
核磁测试:将25 mg 测试样品溶解在1.5 mL 氘代DMSO 中,采 用Bruker 公 司 的Avance 600MHz 核 磁共振仪测试得到核磁共振氢谱(1H NMR)和碳谱(13C NMR)。
液相色谱测试:采用Agilent 1260 高效液相色谱仪对样品纯度分析,色谱柱ZORBAX SB-C18 4.6×250 mm,柱温35 ℃,检测波长320 nm,进样量10 μL,流速1.0 mL·min-1,流动相为乙腈/水=5/95,运行时间10 min,通过面积归一化法计算化合物纯度。
TG-DSC 测试:采用德国耐驰公司的STA449 TG-DSC 联用热分析仪,检测氛围为氮气,升温速率为10 ℃·min-1,测试温度范围为50~400 ℃。
粉末XRD 测试:采用德国Bruker 的D8 Advance型粉末衍射仪,电压40 kV,电流30 mA,2θ测试角度5°~50°,Cu-Kα 射线。
形貌分析:采用德国ZEISS 公司的Scope A1 光学显微镜对载玻片上分散的样品颗粒进行观察。
红外测试:采用Perkin Elmer 傅里叶变换红外吸收光谱仪,通过溴化钾压片测试,测试范围400~4000 cm-1。
元素分析:采用德国Elementar 元素分析仪。
机械感度测试[21]:使用BAM 撞击感度测试仪和BAM 摩擦感度测试仪分别测试样品的撞击感度与摩擦感度,撞感落锤质量10 kg,摩感载荷360 N。
2 结果与讨论
2.1 合成工艺研究
NTO 连续流合成是由TO 在硝酸条件下,利用连续流装置实现目标化合物的连续化合成输出,合成路线如图2 所示。由于TO 固体加入硝酸溶解时会明显放热,为提高进料效率,参考文献[3]将TO 与HNO3按照n(TO)∶n(HNO3)=1∶6 连续加入溶解釜,并同时将溶解液泵入盘管反应器,以确保较低的溶解釜持液量和足够的传热效率。对比不同的硝酸浓度(70%、75%、85%、90%和98%)后发现,当使用85%硝酸时可以较好的兼顾加料速度与溶解液温度,因此以85%硝酸作为硝化体系,研究NTO 连续流合成两个阶段的工艺温度与停留时间对反应收率的影响。NTO 连续流合成中,均相与含固相两个阶段的反应温度分别通过独立的温度控制器控制,停留时间通过改变盘管和管式反应器长度控制。

图2 NTO 连续流合成路线Fig.2 Continuous flow synthesis route of NTO
均相与含固相阶段的停留时间和反应温度对连续流合成NTO 收率的影响如表1 所示。提高均相反应温度可增加反应速率,但当温度超过45 ℃时会有明显气体产生,导致均相反应有效停留时间降低。逐渐延长盘管反应器中均相反应停留时间,反应收率逐渐提高,当均相反应停留时间超过5 min 后,会有固体析出并增加盘管反应器堵塞风险。因此,均相反应阶段最佳硝化温度和停留时间分别为45 ℃和5 min;进一步延长含固相反应阶段停留时间至4 min 后,产率提高到81.4%。继续延长含固相反应阶段停留时间或反应温度,产率均略有下降。这可能是由于继续升高温度或延长反应时间,会导致发生更多副反应,降低反应收率。因此,确定含固相反应阶段的最佳停留时间为4 min,最佳硝化反应温度为45 ℃。

表1 停留时间和反应温度对硝化产率的影响Table 1 Effects of t and T on the yield
2.2 结构与性能表征
2.2.1 结构与纯度
对连续流合成NTO 的化合物结构进行了分析表征,核磁共振NMR:1H NMR(600 MHz,DMSO-d6,25 ℃)δ:13.39 (b,1H),12.83(s,1H);13C NMR(150 MHz,DMSO-d6,25 ℃)δ:154.65,148.19。红外FT-IR(KBr,ν/cm-1):3179(N—H),1682(C=O),1537(—NO2),1329(—NO2),如图3 所示。元素分析(C2H2N4O3,%):Anal.calcd for C 18.46,H 1.54,N 43.08;found C 18.17,H 2.00,N 42.96。结构测试数据与文献[3,7]中NTO 的表征数据一致,确定了连续流合成产物为NTO。
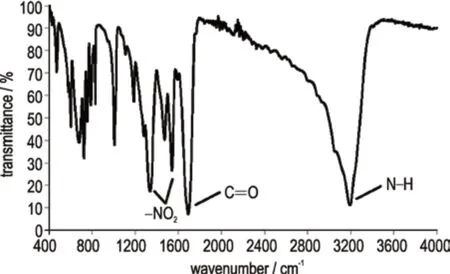
图3 NTO 的FT-TR 谱图Fig.3 FT-IR spectrum of NTO
通过高效液相色谱对连续流合成NTO 纯度进行分析(见图4)。NTO 的色谱保留时间为3.119 min,其他时间无明显杂志峰,按照面积归一化法计算得到NTO 纯度为99.53%。
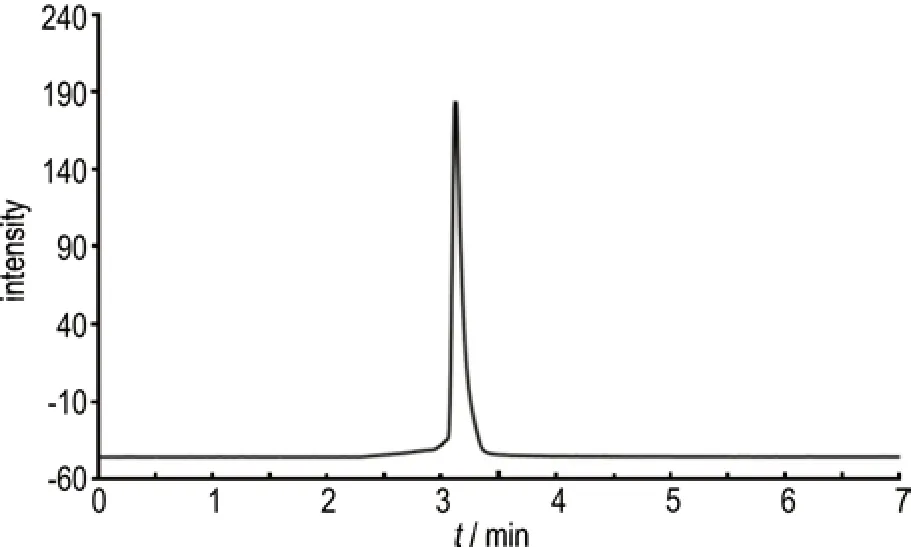
图4 NTO 的高效液相色谱图Fig.4 HPLC spectrum of NTO
2.2.2 晶体形貌
通过光学显微镜对比了釜式合成与连续流合成两种技术路线的产品晶体形貌。如图5a 所示,釜式合成NTO 的晶体粒度分布不均一,呈不规则片状或针状结构,容易发生聚集。釜式合成NTO 的不规则晶体会影响其混合炸药装药,使装药成型性能差、装药密度小、固含量降低,在应用上受到很大的限制[22]。连续流合成NTO 的晶体粒度较细,见图5b。放大显微倍数后发现,NTO 晶体呈块状、缺陷较少且粒度分布较为釜式工艺更加均匀,见图5c~5d。这种晶体形貌差异主要是因为连续流合成工艺具有更高的传质与传热效率。当反应达到一定饱和度开始析晶时,管道内部各单位流体的物料浓度和温度高度一致,使得成核密度及晶体生长环境更加统一,最终得到粒径更小、粒度分布更加均匀的晶体。因此,连续流合成方法更适合制备细颗粒、低缺陷的NTO 晶体。

图5 釜式与连续流合成的NTO 晶体形貌图(a 图为釜式合成NTO,b 图为连续流合成NTO,c~d 为局部放大图)Fig.5 Morphologies of NTO synthesized in flask and continuous flow systems(Fig.a shows NTO by flask method, Fig.b shows NTO by continuous flow method, Fig.c and d show locally enlarged images)
2.2.3 XRD 测试
NTO 具有α和β两种不同的晶型[7],其中α-晶型相对β-晶型更加稳定。对釜式合成与连续流合成两种工艺得到的NTO 分别进行XRD 衍射分析,结果如图6所示。通过对比两种NTO 的XRD 谱图数据可以发现,两者主要衍射峰出现位置均与数据库中α-NTO 卡片一致,说明两种工艺都可直接合成稳定的α-晶型样品。
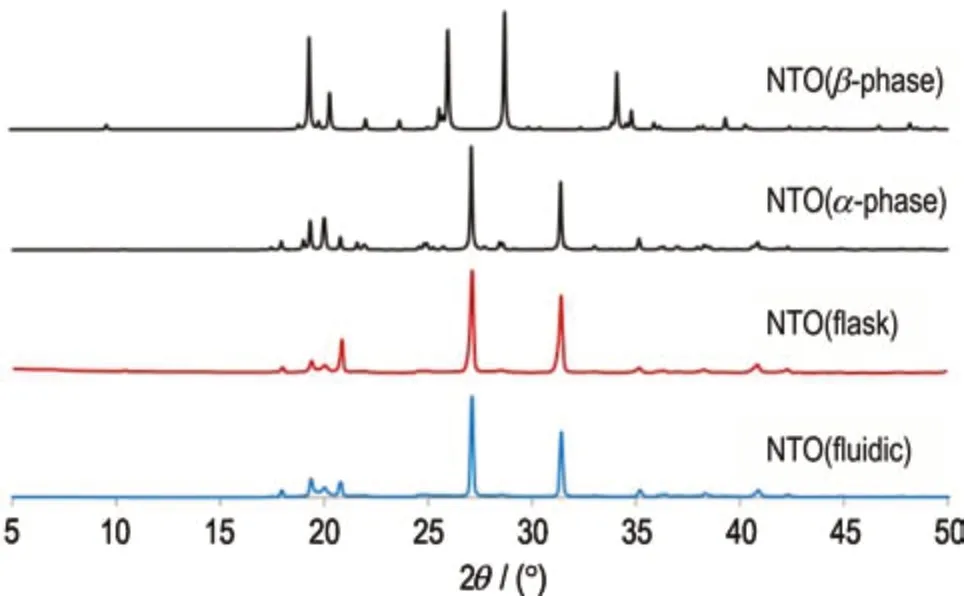
图6 釜式和连续流合成NTO 的XRD 衍射图Fig.6 XRD diffraction patterns of NTO synthesized by flask and continuous flow system
2.2.4 热性能分析
对两种合成工艺得到的NTO 进行热性能分析,TG-DSC 曲线如图7 所示。从TG 曲线可以看出,釜式合成NTO(红色曲线)在173.68~275.06 ℃范围内出现热质量损失,过程的质量损失为80.68%。连续流合成NTO(蓝色曲线)在197.50~278.19 ℃范围内出现热质量损失,过程的质量损失为85.12%。可以看出,连续流合成NTO 的质量损失较釜式NTO 更晚发生,但质量损失比釜式提高4.44%且损失速度更大;从DSC 曲线可以看出,两种合成方法得到的NTO 均先后经历了吸热熔化和放热分解两个阶段。其中,釜式合成NTO 的热分解峰为273.28 ℃,较连续流合成NTO 的热分解温度276.23 ℃略微提前2.95 ℃,这与TG 体现出的热性能趋势基本一致。热性能分析结果说明连续流合成NTO 比釜式合成NTO 具有更好的热稳定性,同时能够发生更充分和更迅速的热分解反应。
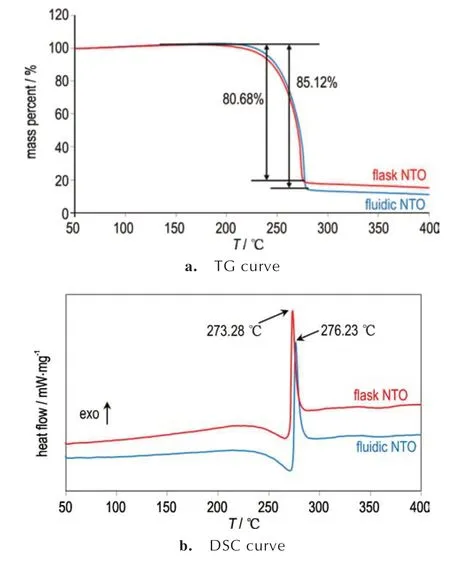
图7 釜式与连续流合成NTO 的TG-DSC 图Fig.7 TG-DTG curves of NTO synthesized in flask and continuous flow system
2.2.5 机械感度表征
通过BAM 感度仪对两种合成工艺得到的NTO 进行了机械感度测试。结果发现,连续流合成NTO 在40 J 和360 N 的撞击感度与摩擦感度测试中均未发火,与釜式合成NTO 的机械感度测试结果一致,说明连续流直接合成的NTO 保持了与传统工艺NTO 一样的优良安全性能。连续流合成NTO 的晶体粒度较小,比表面积更大且粒度分布更加均匀,可以较好吸收、传导外界刺激产生的热点能量,减少热量累积造成的化合物分解。
2.3 反应效率与收率
与釜式合成工艺[6]对比,连续流合成工艺的传质效率更快,同时对反应温度的控制更加准确,可以加速硝化反应的表观反应动力学过程,同时降低副反应发生。如图8 所示,NTO 的连续流合成反应停留时间仅为9 min,较釜式反应停留时间(1.5~2.0 h)显著缩短,提高了反应效率。在相同的产能前提下,连续流合成工艺的在线持药量较釜式可降低为原来的1/12,同时连续流反应收率(81.4%)与釜式合成收率(78.0%)相当。

图8 连续流合成工艺优点Fig.8 Advantages of the continuous flow synthesis
3 结 论
本研究根据NTO 不同反应阶段的体系特点,开展其连续流合成工艺研究,对比了两种合成工艺的样品晶型、形貌、热性能、机械感度以及合成效率,得到结论如下:
(1)以85%硝酸溶液作为硝化体系,在n(TO)∶n(HNO3)=1∶6,硝 化 温 度 为45 ℃,反 应 液 流 速 为14.4 mL·min-1,均相硝化反应停留时间5 min,含固相硝化反应停留时间4 min 的连续流合成工艺条件下,以81.4%的收率合成了纯度为99.53%的NTO 样品。
(2)连续流合成工艺得到的NTO 晶体呈块状、缺陷较少,样品为α-晶型,热分解温度276.23 ℃,热分解过程质量损失为85.12%,撞击感度IS>40 J,摩擦感度FS>360 N。较釜式工艺而言,晶体粒度分布更加均匀,热分解峰温延后2.95 ℃,质量损失增加4.44%,安全性能保持不变。
(3)针对NTO 釜式合成工艺存在的安全风险,开发了一种安全、高效的NTO 连续流合成工艺,分段设计装置具有较好的防堵塞能力,显著降低了硝化反应停留时间和在线药量。
猜你喜欢
环境保护与循环经济(2017年7期)2018-01-22
中国环境科学(2016年3期)2016-02-08
水资源保护(2015年5期)2016-01-07
企业导报(2015年20期)2015-11-30
科技与创新(2015年10期)2015-07-07
应用海洋学学报(2014年2期)2014-11-26
应用化工(2014年7期)2014-08-09
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
中国医学影像学杂志(2013年12期)2013-05-06
