
O-季铵盐氧化壳聚糖的合成及对棉织物的抗菌整理
2024-03-25 02:41周晓龙汪瑞琪陈国丽郭文明孙立德汤大保许云辉
高等学校化学学报 2024年3期
周晓龙,汪瑞琪,陈国丽,郭文明,陈 飞,周 炎,孙立德,汤大保,许云辉
(1. 安徽农业大学材料与化学学院, 合肥 230036; 2. 安徽京九丝绸股份有限公司, 阜阳 236000)
壳聚糖是自然界中唯一已知的天然碱性多糖,因其来源丰富、 可再生、 独特的生理及生物活性而被广泛应用[1,2]. 但壳聚糖分子间较强的氢键作用使其难溶于水,且其抗菌性受到分子量、 脱乙酰度和pH值的影响,因而限制了其应用范围[3,4]. 在壳聚糖中引入位阻大、 水合能力强的季铵盐基团能削弱分子间氢键,提高壳聚糖的水溶性及抑菌性. 目前壳聚糖季铵化研究已成为壳聚糖化学改性的重要方向[5,6].
近年来,在壳聚糖分子C2位氨基上进行季铵化或接枝季铵盐引起很多学者的关注. Yin等[7]首先使用二甲氨基氯乙烷盐酸盐以及5,5-二甲基海因制备了(5,5-二甲基苯乙烯基)-3-乙基二甲胺(ENDMH),然后将ENDMH与壳聚糖在80 ℃下反应24 h制备了季铵盐壳聚糖. Ke等[8]使用3-氯-2-羟丙基三甲基氯化铵,通过乳液交联反应对壳聚糖C2位的—NH2基进行取代,获得N-季铵化壳聚糖产物. 也有学者在壳聚糖分子的—NH2上先形成席夫碱,再将席夫碱还原,然后与卤代烃反应后转化为季铵盐,此方法可以在壳聚糖分子中引入不同长度的烷基链[9,10]. 但这些方法反应周期长,使用的化学助剂对人体和环境存在危害,应用受限,且在反应过程中会消耗大量的壳聚糖氨基,使壳聚糖的天然抑菌性和阳离子特性被破坏[11,12].
为了保护壳聚糖的C2位氨基和天然性质,许多学者先在壳聚糖中的氨基上形成席夫碱进行保护,然后在C6位羟基上接枝季铵盐,再脱除保护基团,获得O-季铵化壳聚糖产物. 高艳丽等[13]利用甲壳素分子中的C6位羟基的反应活性大于C2位乙酰氨基和C3位仲羟基的特性,在水体系中将α和β两种不同晶型的甲壳素与环氧丙基三甲基氯化铵(GTMAC)发生C6位亲核取代,合成出C6位取代的O-甲壳素季铵盐衍生物. 刘新等[14]和林友文等[15]均采用苯甲醛与壳聚糖先在C2位发生席夫碱反应,再在C6位接枝2,3-环氧丙基三甲基氯化铵(GTA)后脱保护,合成出O-季铵盐壳聚糖. 但苯甲醛化学性质不稳定,有一定毒性,且该类季铵盐壳聚糖分子中的反应基团较少,与纺织纤维缺少有效的化学键结合,耐水洗性能差.
香草醛被称为“食品香料之王”,安全无毒,应用面广,也常被用于合成香草醛-壳聚糖席夫碱[16]及香草醛-壳聚糖季铵盐[17]等壳聚糖衍生物. 为了增强壳聚糖衍生物与纤维材料的结合牢度,一些学者采用高碘酸钠选择性氧化壳聚糖制备出双醛基壳聚糖[18],并使其分别与棉织物和羊毛形成半缩醛和席夫碱等交联[19,20],从而实现纤维材料的持久抗菌和低盐染色等性能.
本文采用香草醛与壳聚糖反应形成席夫碱保护C2 位氨基,再在壳聚糖的C6 位进行取代后脱去席夫碱,合成了O-季铵盐壳聚糖(O-HACC),然后使用KIO4将O-HACC中C2位和C3位的部分基团选择性氧化为醛基,得到了新型的O-季铵盐氧化壳聚糖(O-HAOCC). 由于在C6 位接枝了季铵盐基团,O-HAOCC的水溶性好,且保持了壳聚糖的聚阳离子氨基的抑菌活性,具有季铵盐与阳离子氨基的双重抗菌能力; 同时O-HAOCC 分子中的双醛基团可与纺织纤维形成半缩醛及席夫碱等化学键合,可广泛用于持久抗菌的纤维材料、 纺织服装、 家纺面料和生物医疗等领域,拓展了壳聚糖的应用范围,为合成功能性壳聚糖衍生物与研发抗菌生态纺织品开辟了新途径.
1 实验部分
1.1 试剂与仪器
壳聚糖(CTS),脱乙酰度≥95%,黏均分子量为7.2×105,化学纯,西格玛奥德里奇(上海)贸易有限公司; 2,3-环氧丙基三甲基氯化铵(GTA)和高碘酸钾(KIO4),分析纯,上海阿达玛斯贝塔化学试剂有限公司; 香草醛、 过硫酸钾和丙酮,分析纯,上海阿拉丁生化科技股份有限公司; 2,2'-联氮双(3-乙基苯并咪唑-6-磺酸)二铵盐(ABTS),分析纯,上海麦克林生化科技股份有限公司; 乙酸和无水乙醇,分析纯,上海泰坦科技股份有限公司; 纯棉织物(118 g/m2),绍兴丰锦纺织品有限公司; 大肠杆菌(E. coli,ATCC 8099)和金黄色葡萄球菌(S. aureus,ATCC 6538),安徽医科大学.
Tensor Ⅱ型傅里叶变换红外光谱仪(FTIR),在400~4000 cm-1范围进行扫描,分辨率为4 cm-1,德国Bruker公司; MXPAHF型X射线衍射仪(XRD),管电压为36 kV,管电流为20 mA,扫描速度为4°/min,2θ范围为5°~50°,日本玛珂公司; Agilent DD2 600 MHz 型超导傅里叶变换液体核磁共振波谱仪(1H NMR),瑞士Bruker 公司; Mettler Toledo 型热重分析仪(TGA),瑞士梅特勒托利多集团; Hitachi S-4800型扫描电子显微镜(SEM),日本日立公司; UV-4802S型紫外分光光度计,上海尤尼柯仪器有限公司; KDN-B型全自动凯氏定氮仪,上海欣嘉科技有限公司.
1.2 O-季铵盐壳聚糖的合成
1.2.1 壳聚糖席夫碱(Schiff-CTS)的合成 25 ℃下,将2 g 壳聚糖溶于120 mL 体积分数为10%的乙酸中,加入40 mL无水乙醇,在搅拌下于30 min内滴加含有14.6 g香草醛的乙醇溶液,然后于60 ℃加热回流20 h,冷却后在高速搅拌下滴加0.5 mol/L NaOH溶液调节pH值为7.0,将析出的沉淀抽滤后用甲醇充分超声洗涤,于50 ℃真空干燥后得到Schiff-CTS粉末(Scheme 1).

Scheme 1 Synthetic route of O-HAOCC
1.2.2O-季铵盐席夫碱壳聚糖(Schiff-HACC)的合成 将Schiff-CTS 置于三口烧瓶中,加入40 mL 质量分数为0.1%~0.5%的NaOH溶液,搅拌使其分散均匀[16],然后按照一定比例(Schiff-CTS与GTA摩尔比分别为2∶1,1∶1,1∶2,1∶3,1∶4 和1∶5)分3 次(每15 min 加1 次)加入GTA,在40~90 ℃下搅拌反应1~7 h,得到黏稠状产物,用无水乙醇沉淀、 过滤,再用80%的乙醇溶液超声洗涤30 min,所得粗产物在无水乙醇为溶剂的索氏萃取器中萃取24 h,然后于70 ℃真空干燥,即得到Schiff-HACC 固体粉末(Scheme 1). 最优的合成条件为NaOH 质量浓度0.2%,Schiff-CTS 与GTA 摩尔比1∶3,反应温度80 ℃,反应时间3 h; 将最优条件下制备的产物命名为Schiff-HACC-0.2-1/3-80-3.
1.2.3O-季铵盐壳聚糖(O-HACC)的合成 将Schiff-HACC 置于50 mL 0.25 mol/L 的HCl 乙醇溶液中,室温搅拌24 h后蒸发去除大部分乙醇,得到胶状物,加入15 mL去离子水溶解,再用丙酮沉淀、 抽滤,80 ℃真空干燥后获得粉末状固体,即为O-HACC(Scheme 1),收率为63.75%~88.62%. 将利用最优条件下制备的Schiff-HACC合成的产物命名为O-HACC-0.2-1/3-80-3.
1.3 O-季铵盐氧化壳聚糖的制备
将O-HACC-0.2-1/3-80-3按浴比[O-HACC质量(g)与去离子水体积(mL)比]1∶50溶于去离子水中,用体积分数为10%的乙酸溶液调节pH值为4.0,加入不同质量的高碘酸钾使溶液中的氧化剂浓度分别为4 g/L和8 g/L,在25 ℃下避光搅拌反应1~8 h后,添加2倍体积的无水乙醇终止反应,然后将反应液装入透析袋中流水透析2~3 d,所得溶液经60 ℃旋转蒸发,得到O-季铵盐氧化壳聚糖(O-HAOCC)固体粉末(Scheme 1). 根据氧化剂浓度和反应时间,命名为O-HAOCC-x-y[x(g/mL)为KIO4浓度,取值为4和8;y(h)为反应时间,取值为1~8].
1.4 O-季铵盐氧化壳聚糖改性棉织物的制备
将煮练退浆的棉织物浸渍在质量分数为0.5%~4.0%的O-HAOCC(由O-HACC-3-80用4 g/L KIO4溶液氧化3 h制得的样品)溶液中,使用稀盐酸调节反应液的pH值为4.0左右,在80 ℃恒温水浴中搅拌反应2 h. 反应结束后取出棉织物,放入真空烘箱中于60 ℃预烘20 min,然后升温至80 ℃干燥2 h,再将干燥后的棉织物用清水浸泡8 h,脱水、 晾干后,即得到不同接枝率的O-HAOCC改性棉织物.
1.5 O-HACC季铵盐取代度的测定
参照文献[21]方法测定O-HACC 的季铵盐取代度(DS). 将0.2 gO-HACC 季铵盐用去离子水定容至50 mL,移取25 mL样品溶液,加入25 mL去离子水,然后用0.1 mol/L 的氢氧化钠溶液调节pH值在8.6~9.2之间,滴加1 mL质量分数为8%的铬酸钾指示剂,使用0.05 mol/L的硝酸银进行滴定.
式中:c(mol/L)为硝酸银溶液浓度;V(mL)为消耗的硝酸银溶液的体积;m(g)为被滴定的O-HACC的质量;n0为O-HACC的单元分子量,本文中为297;n1为原壳聚糖的单元分子量,本文中为163.
1.6 O-HAOCC氧化度的测定
实验中使用KIO4将O-HACC中C2位和C3位的部分基团选择性氧化为醛基,利用紫外-可见分光光度计法测试O-HAOCC中的醛基含量[22]. 在最大吸收波长245 nm处测试一系列浓度的高碘酸钾溶液吸光度值,并绘制出标准曲线,然后测试氧化反应结束后高碘酸钾溶液的吸光度并计算其浓度,通过下式计算O-HAOCC的氧化度(DO,mmol/g):
式中:c0(mg/mL)为反应前高碘酸钾的浓度;c1(mg/mL)为反应结束后高碘酸钾的浓度;V(mL)为反应液的体积;M(230)为高碘酸钾的摩尔质量;m(g)为加入的O-HACC干燥质量.
1.7 O-HAOCC收率的测定
产物收率能一定程度上反映O-HAOCC的氧化降解程度,计算公式如下:
式中:Y(%)为O-HAOCC收率;m2(g)为反应前加入反应物O-HACC的干燥质量;m1(g)为反应结束之后回收产物O-HAOCC的干燥质量;M1(298.46)为原料O-HACC葡萄糖单元的摩尔质量;M2(326.47)为产物O-HAOCC葡萄糖单元的摩尔质量.
1.8 水溶性测定
将0.05 g 样品在室温下搅拌溶解在10 mL 去离子水中,当加入的样品完全溶解后,再每次加入0.05 g样品,直至搅拌样品不再溶解为止. 离心除去未溶解的样品,按下式计算样品在水中的溶解度(S,g/L):
式中:m0(g)为加入水中的样品质量;m'(g)为未溶解的样品质量;V(L)为去离子水的体积.
1.9 黏均分子量测定
将O-HACC或O-HAOCC溶解在0.2 mol/L NaCl+0.1 mol/L CH3COOH溶液中,配制成2.0 mg/mL的溶液. 采用黏度法测量并计算出其在(25.0±0.1) ℃下的特性黏度[η],从而计算其黏均分子量(Mv)[23]. 当采用黏度法测试的样品是高分子的稀溶液时,常数K和α值的变化对检测结果影响较小.测试发现,当α在0.8~1.0区间变化时,壳聚糖黏均分子量误差在4%以内. 溶液的特性黏度与黏均分子量之间满足如下关系式:
1.10 抗氧化性测定
配制浓度为2.6 mmol/L 的K2S2O8溶液与浓度为7.4 mmol/L 的ABTS·+溶液,分别取2 mL混合均匀,于25 ℃下避光反应12 h,制得ABTS·+自由基溶液,再用无水乙醇稀释ABTS·+自由基溶液(稀释约53倍),使其在734 nm处的吸光度为(0.7±0.025)[24]. 将2 mgO-HACC或O-HAOCC溶于2 mL去离子水中,再与8 mL 稀释后的ABTS·+溶液发生反应,然后每5 min 测定溶液在734 nm 处的吸光度,共测试8次. 另取2 mL去离子水与8 mL稀释后的ABTS·+溶液混合均匀,作为空白样. 利用下式计算自由基消除率(R,%),R越高,则抗氧化性能越强.
式中:A0为空白样的吸光度;A1为样品反应后的吸光度.
1.11 改性棉织物上O-HAOCC的接枝率测定
棉织物中不含N元素,而O-HAOCC中含有较多的N元素,使用凯氏定氮法测定O-HAOCC改性棉织物上N元素含量,计算公式如下:
式中:N(%)为样品中N 元素含量;VT和VB(mL)分别为样品和空白滴定中使用的盐酸溶液的体积;M(mol/L)为标准盐酸溶液的浓度;mf(mg)为测试样品的干燥质量.
改性棉织物上的O-HAOCC接枝率(G,%)按下式计算:
式中:mf(g)为改性棉织物的干燥质量.
1.12 改性棉织物的服用性能测定
棉织物的力学性能、 吸水毛细效应、 白度和折皱回复角分别按照相关标准GB/T 3923.1-2013《纺织品织物拉伸性能》(样品夹持长度15 cm、 拉伸速度200 mm/min)、 FZ/T 01071-2008《纺织品毛细效应试验方法》、 GB/T 17644-2008《纺织纤维白度色度试验方法》和GB/T 3819-1997《纺织品织物折痕回复性的测定》进行测试.
1.13 改性棉织物的抗菌性能测定
参照美国AATCC Test Method 100-1999《定量测试方法》对空白棉织物或O-HAOCC改性棉织物进行抗菌性能测试. 根据下式计算织物的抗菌率(D,%):
式中:C0和C分别为空白棉织物和改性棉织物的细菌菌落数. 抗菌耐久性测试参照FZ/T 73023-2006《抗菌针织品附录C: 抗菌织物试样洗涤试验方法》进行50次洗涤测试.
2 结果与讨论
2.1 O-HACC和O-HAOCC的制备条件分析
壳聚糖氨基中带有孤电子对的氮原子可与香草醛的醛基上的碳原子发生亲核加成反应形成壳聚糖席夫碱(Shciff-CTS). 由于线型大分子壳聚糖存在较大的位阻影响,本文采用过量的香草醛与壳聚糖进行均相反应使壳聚糖C2 位氨基全部生成席夫碱. 理论上壳聚糖C2 位的—NH2,C3 位和C6 位的—OH均为亲核基团,其中C2位—NH2供电子性最强,与香草醛形成席夫碱受到保护; C3位—OH 为仲醇羟基,不活泼,且位于吡喃糖环内,空间位阻大,亲核取代反应难发生[25]. 吡喃糖环外的C6位—OH为伯羟基,位阻小,活性较高,故Schiff-CTS与GTA的取代反应主要发生在壳聚糖的C6位—OH,生成了C6位取代的O-季铵盐壳聚糖.
DS是衡量壳聚糖C6位季铵盐基团接枝程度的物理量,图1示出了反应条件对壳聚糖季铵盐取代度的影响. 由图1可见,Schiff-CTS与GTA在碱性条件下发生亲核取代反应,季铵盐取代度随着GTA用量的增加先增大后减小,Schiff-CTS 与GTA 的摩尔比为1∶3 时的取代度达到最高值(86.35%). 因为Schiff-CTS与GTA的反应是非均相反应,随着GTA用量增多,与壳聚糖接触的几率增大,促进亲核取代反应的进行; 但继续提高GTA 的用量,由于Schiff-CTS 中可供取代的基团数量有限,同时较大的GTA季铵盐基团的引入使壳聚糖空间位阻增大,取代反应不易继续进行,且过多的GTA导致GTA自身的副反应增多[26],造成取代度降低. 反应温度对季铵盐取代反应影响较大,随着反应温度升高,Schiff-CTS的季铵盐取代度明显增加,在80 ℃时,取代度最大. 提高温度可加快反应物的动能,有利于GTA分子克服在Schiff-CTS表面的空间位阻,从而提高产物取代度; 但温度过高会引起Schiff-CTS的主链降解和GTA的环氧键断裂形成交联,不利于Schiff-CTS和GTA的取代反应进行.
Schiff-CTS 的结构中有多个可能发生取代反应的羟基,其中C6 位的一级醇羟基和香草醛的酚羟基在碱性条件下可分别形成氧负离子和酚氧负离子,更容易发生取代反应生成Schiff-HACC(Scheme 1),而C3 位的二级羟基位阻大,难以发生反应,因此理论上Schiff-CTS 中接枝GTA 季铵盐的位置不是唯一的,而是多种羟基的竞争反应[15]; 同时NaOH溶液能充分溶胀Schiff-CTS,促使壳聚糖中的活性基团显露出来,增加与GTA 接触的几率,这均有利于Schiff-CTS 与GTA 进行亲核取代反应,生成O-季铵盐席夫碱壳聚糖(Schiff-HACC). 由图1可见,中性环境下Schiff-CTS的季铵盐取代度较低,不足65%,而加入催化剂NaOH溶液显著提高了取代度,当NaOH质量分数为0.2%时,Schiff-CTS的取代度达到86%以上. 实验中发现,碱浓度增大,反应体系澄清速度加快,表明碱性条件促进了取代反应的发生; 而当碱浓度过高时,GTA的环氧键易断裂,形成邻羟基[25,26],反应活性降低,导致产物取代度减小. 由图1可知,延长反应时间,Schiff-CTS中C6位取代度逐渐提高,反应3 h的取代度达到最高; 但反应时间继续延长,产物的季铵盐取代度反而降低. 这是由于取代反应时间延长使Schiff-CTS的接枝链不断增长,反应体系黏度增大,不易与GTA接触反应,且反应时间延长易引起GTA水解,造成两者的取代反应效率降低.
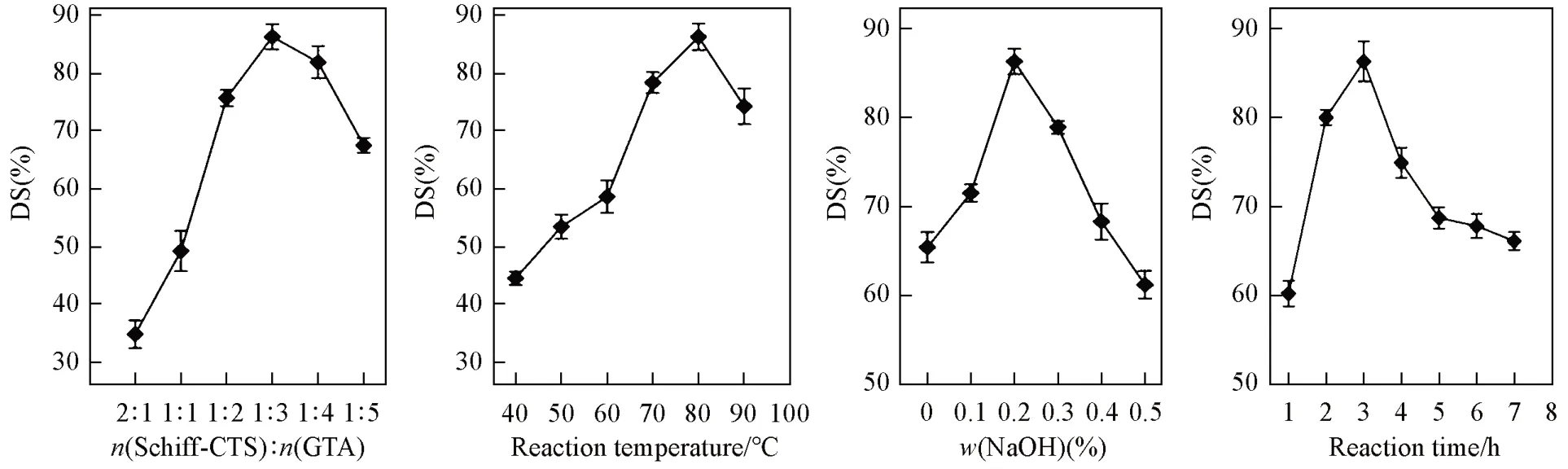
Fig.1 Effects of different reaction conditions on the degree of substitution of quaternary ammonium salt for chitosan(A) w(NaOH)=0.2%, reaction temperature was 80 ℃, reaction time was 3 h; (B) w(NaOH)=0.2%, n(Schiff-CTS)∶n(GTA)=1∶3,reaction time was 3 h; (C) n(Schiff-CTS)∶n(GTA)=1∶3, reaction temperature was 80 ℃, reaction time was 3 h; (D) w(NaOH)=0.2%, n(Schiff-CTS)∶n(GTA)=1∶3, reaction temperature was 80 ℃.
高碘酸钾在氧化O-HACC的过程中能劈断季铵盐壳聚糖的吡喃糖环中的C2—C3键[18~20],使C2位和C3位上相邻的氨基和羟基选择性氧化成醛基,得到双醛基季铵盐氧化壳聚糖(O-HAOCC). 由图2可知,随着氧化时间的延长,不同浓度氧化剂处理的O-HAOCC的醛基含量逐渐增多,而氧化产物收率不断减少,且相同时间下8 g/L 的KIO4氧化的季铵盐壳聚糖的醛基含量明显高于4 g/L 的KIO4氧化的产物,但其氧化产物的收率下降较大,8 g/L KIO4氧化3 h 的O-HAOCC 收率已不到74%,而4 g/L KIO4氧化3 h的产物收率较高(88.67%). 这是由于KIO4选择性氧化反应中,季铵盐壳聚糖的葡萄糖苷键会发生氧化降解等副反应[18],从而对产物收率产生显著影响,增加氧化剂浓度及氧化时间,季铵盐壳聚糖的降解反应加快,产物收率降低严重. 因此,选择4 g/L的KIO4对O-HACC进行局部有限氧化,氧化时间在1~3 h,可得到醛基含量为0.134~0.692 mmol/g(每100 个O-HACC 葡萄糖单元中有2~10 个被氧化)、 产物收率为88.67%~97.21%的O-HAOCC.O-HAOCC分子中的活性醛基在酸性条件下可与纤维素纤维的羟基[16,19]或蛋白质纤维的氨基[20]分别发生交联反应,能用于纤维材料的持久抗菌,且O-HAOCC中仍含有大量的阳离子氨基,保持了壳聚糖的天然抑菌性,故O-HAOCC具有季铵盐与阳离子氨基的双重抗菌活性.

Fig.2 Effects of oxidation concentration and time of KIO4 on the degree of oxidation and yield of O-HAOCCThe O-HACC used was O-HACC-0.2-1/3-80-3.
2.2 结构与形貌表征
图3 为O-HAOCC 合成过程中每个阶段产物的FTIR 谱图. 与CTS 的红外谱图相比,Schiff-CTS 和Schiff-HACC分别在724.1,763.2及1580.8 cm-1处出现芳环邻二取代的特征双吸收峰和苯环骨架伸缩振动峰,且在1644.6 cm-1处存在γC=N的强吸收峰,说明香草醛已在壳聚糖的C2位形成了席夫碱结合.而Schiff-HACC 和O-HACC 均在1518.4 cm-1附近出现了明显的季铵盐基团甲基的δC—H特征峰,表明在壳聚糖C6 位上成功接枝了季铵盐侧链. 同时,O-HACC 红外谱线中的苯环特征峰全部消失,位于1644 cm-1处的席夫碱吸收带显著减弱,表明Schiff-HACC经盐酸乙醇溶液处理后脱除了香草醛苯环,且在1518.4 和1590.6 cm-1处分别出现对应季铵盐基团与酰胺II 的βN—H振动峰,这进一步证明席夫碱能有效保护CTS 的C2 位氨基并方便去除保护基,从而在壳聚糖的C6 位上引入季铵盐. 此外,图3 显示O-HAOCC在1737.2和2846.1 cm-1位置分别存在归属于醛基的νC=O和νC—H振动峰,且在895.2 cm-1处出现半缩醛特征谱带,同时季铵盐特征峰偏移到1520.3 cm-1附近,说明KIO4选择性氧化已在O-HAOCC分子中产生活性醛基.
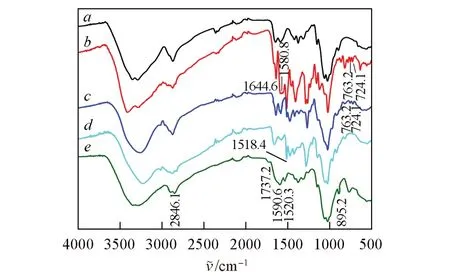
Fig.3 FTIR spectra of products at each synthesis stage of O-HAOCCa. CTS; b. Schiff-CTS; c. Schiff-HACC-0.2-1/3-80-3;d. O-HACC-0.2-1/3-80-3; e. O-HAOCC-4-3.
通过XRD 研究CTS,O-HACC 与O-HAOCC 的物相结构. 由图4 可以看出,CTS 在2θ=10.78°和20.11°位置出现2 个明显的衍射峰,分别归属于壳聚糖结晶形态I和Ⅱ的(100)晶面[14]. 在O-HACC 的XRD谱中,2θ=10°附近的衍射峰已基本消失,同时2θ=20°附近的衍射峰强度下降较大,表明壳聚糖两种晶型中分子链的规整程度不同,其中结晶态I更易遭到破坏,且壳聚糖的C6位羟基接枝季铵盐基团后,侧链的位阻增大,形成氢键的能力降低,导致一部分结晶型转变成无定形. 而O-HAOCC在2θ=10°附近的衍射峰完全消失,且在2θ=20°附近的衍射峰也减弱并且宽化,这说明KIO4选择性氧化使OHACC的C2—C3键断裂发生开环反应[18],削弱了壳聚糖分子间的氢键作用,造成O-HAOCC 的晶体结构破坏和内部结构疏松,即表现出O-HAOCC水溶性极大提高.
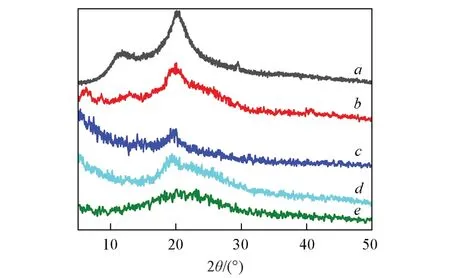
Fig.4 XRD patterns of products at each synthesis stage of O-HAOCCa. CTS; b. Schiff-CTS; c. Schiff-HACC-0.2-1/3-80-3;d. O-HACC-0.2-1/3-80-3; e. O-HAOCC-4-3.
图5 为壳聚糖及其衍生物的1H NMR 谱图. 在δ2.98 处为CTS 上与氨基相连的C2 上的质子峰,δ3.45~3.90处为CTS糖环上C3,C4,C5和C6的质子峰.O-HACC在δ3.14附近出现了明显的强峰,归属于季铵盐基团—N+(CH3)3,且在δ2.05处出现了C2位的—NH2质子峰,表明通过席夫碱保护CTS的C2位氨基,在其C6位—OH上引入了GTA侧链; 同时O-HACC在δ7.42和7.23处香草醛苯环的质子峰消失,说明实验过程中HCl乙醇溶液已将Schiff-HACC中香草醛在壳聚糖C2位形成的席夫碱保护完全脱除. 而经KIO4氧化反应后,O-HAOCC 在δ2.05 处—NH2质子峰和δ2.98 处C2 位质子峰的峰面积均有较大幅度下降,这是由于O-HACC分子中C2和C3位上的部分氨基、 羟基被KIO4选择性氧化; 此外,与O-HACC 的1H NMR 谱图比较,O-HAOCC 在δ2.48,2.73,3.14 和4.23 附近对应于季铵盐基团中各碳位上的质子峰发生偏移,可能是因为氧化后的O-HAOCC 分子中的化学环境改变所致,这也被红外分析所证实.
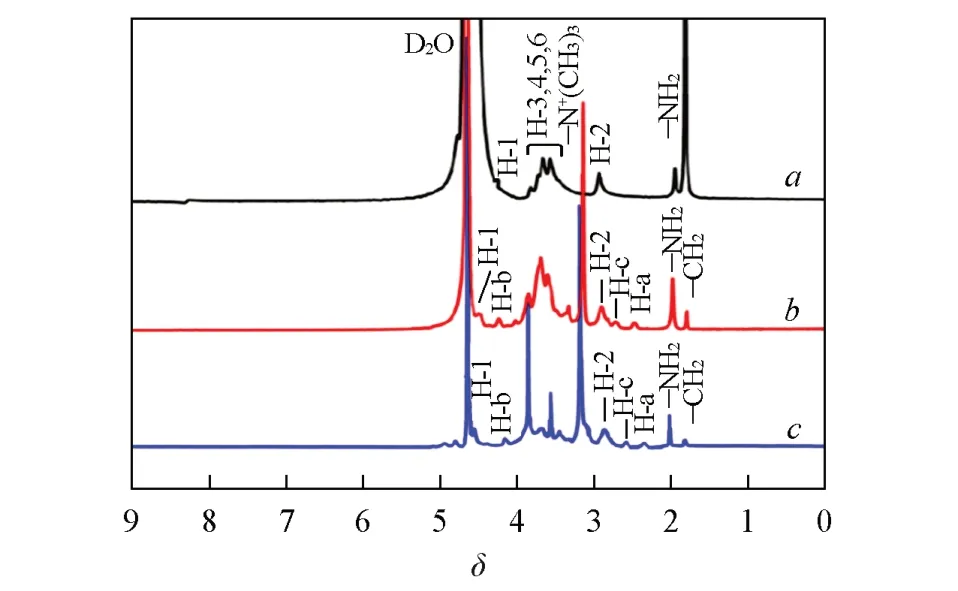
Fig.5 1H NMR spectra of CTS(a), O-HACC-0.2-1/3-80-3(b) and O-HAOCC-4-3(c)
图6示出了CTS,O-HACC和O-HAOCC的DSC曲线. 所有样品均在68~125 ℃范围存在一个较宽的吸热峰,主要与样品中结合水的蒸发有关. 由图6谱线b~f可知,O-HACC和O-HAOCC的水吸热峰面积比CTS 增大,表明引入亲水性的季铵盐基团和醛基均可增强壳聚糖对水分子的吸附能力. 壳聚糖的DSC曲线在272~360 ℃区间有一个较强的分解放热峰,反映了CTS的热稳定性能. 随着壳聚糖季铵盐取代度从0增加到86.35%,O-HACC的放热吸收峰从303.4 ℃逐渐降低到206.8 ℃,且放热峰面积也减小,这是因为较大位阻的GTA分子接枝到壳聚糖C6位,造成O-HACC分子间氢键力的破坏,减少了分子结晶形态,这与XRD 分析结果相一致. 另外,随着KIO4氧化时间延长,O-HAOCC 的DSC 曲线e和f中的吸收峰起伏较多,热分解模型变得复杂,放热峰位置进一步下降到198.5 ℃(氧化3 h)和186.2 ℃(氧化6 h),说明季铵盐壳聚糖氧化中发生了吡喃糖环的开环反应,其结晶结构被削弱,同时氧化降解副反应导致O-HAOCC的分子量减小和分子间氢键结合减弱,从而使氧化产物的热分解能和热稳定性降低.
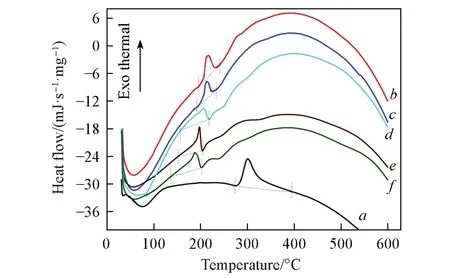
Fig.6 DSC curves of CTS(a), O-HACC-0.2-1/3-40-3(b)and O-HACC-0.2-1/3-80-3(c), O-HAOCC-4-1(d),O-HAOCC-4-3(e) and O-HAOCC-4-6(f)
图7 为CTS,O-HACC 与O-HAOCC 的SEM 照片. 大分子CTS 是单斜晶系的β-壳聚糖,其表面较光滑,晶型结构完整[图7(A)]. 随着季铵盐取代度的提高,O-HACC 的表面结构变得疏松,出现了较多的空隙结构[图7(B),(C)]. 从图7(D)~(F)可以看出,O-HAOCC颗粒边界逐渐模糊,表面出现结构坍陷,部分区域呈弥散状态; 随着氧化度增大,O-HAOCC表面形成更多的沟壑孔洞结构,颗粒的松散程度增加[图7(F)],这可能是由于KIO4选择性氧化反应破坏季铵盐壳聚糖的结晶形态所致[16,18],这与XRD和热分析结果相符合.

Fig.7 SEM images of CTS(A), O-HACC-0.2-1/3-40-3(B) and O-HACC-0.2-1/3-80-3(C),O-HAOCC-4-1(D), O-HAOCC-4-3(E) and O-HAOCC-4-6(F)
2.3 O-季铵盐氧化壳聚糖性能分析
2.3.1 水溶性和分子量 表1 为不同取代度的O-HACC 和不同氧化度的O-HAOCC 的水溶性及黏均分子量数据. 由表1可以看出,随着取代度提高,在壳聚糖C6位上接枝的亲水性季铵盐基团增多,同时破坏了分子间氢键作用,水分子容易进入,故O-HACC具有良好的水溶性,且接枝季铵盐后的O-HACC分子量有所降低. KIO4选择性氧化的季铵盐壳聚糖黏均分子量下降较大,氧化8 h 的分子量降低到1.04×105,表明氧化时间延长,O-HACC分子链的氧化降解明显加快,但O-HAOCC在水中的溶解度增加,氧化6 h的O-HAOCC水溶性达到279 g/L,这是由于KIO4氧化反应一定程度上破坏了季铵盐壳聚糖的晶体结构[22],O-HAOCC的内部结构更加松散,且分子量减小削弱了氢键结合力,因而水溶性极大提高; 但过度氧化可能产生副产物,导致氧化8 h的O-HAOCC溶解度略有降低.
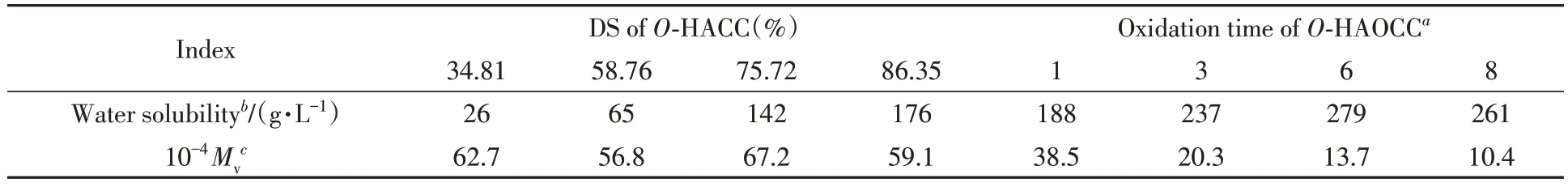
Table 1 Water solubility and molecular weight of O-HACC with different DS and O-HAOCC after oxidation with 4 g/L KIO4 for different time
2.3.2 抗氧化性能 将样品与ABTS·+溶液反应使其褪色,测试734 nm处的特征吸光度值,得到ABTS·+自由基清除率曲线(图8). 反应5 min 时,CTS 的自由基清除率不到5%,抗氧化性弱; 而O-HACC 与O-HAOCC的自由基清除率分别达到44.11%和29.03%~41.15%,具有较强的抗氧化能力. 3种抗氧化剂的ABTS·+自由基清除率均随时间的延长而逐渐增加,但CTS 的抗氧化性能提高缓慢,40 min 的ABTS·+自由基清除率只有18.13%,此时取代度为86.35%的O-HACC和氧化3 h的O-HAOCC的ABTS·+自由基清除率分别高达71.21%和65.58%,ABTS·+反应液溶液明显褪色,紫外吸收峰急剧减弱; 然而氧化时间达到6 h时,O-HAOCC的ABTS·+自由基清除率降低至48.96%.
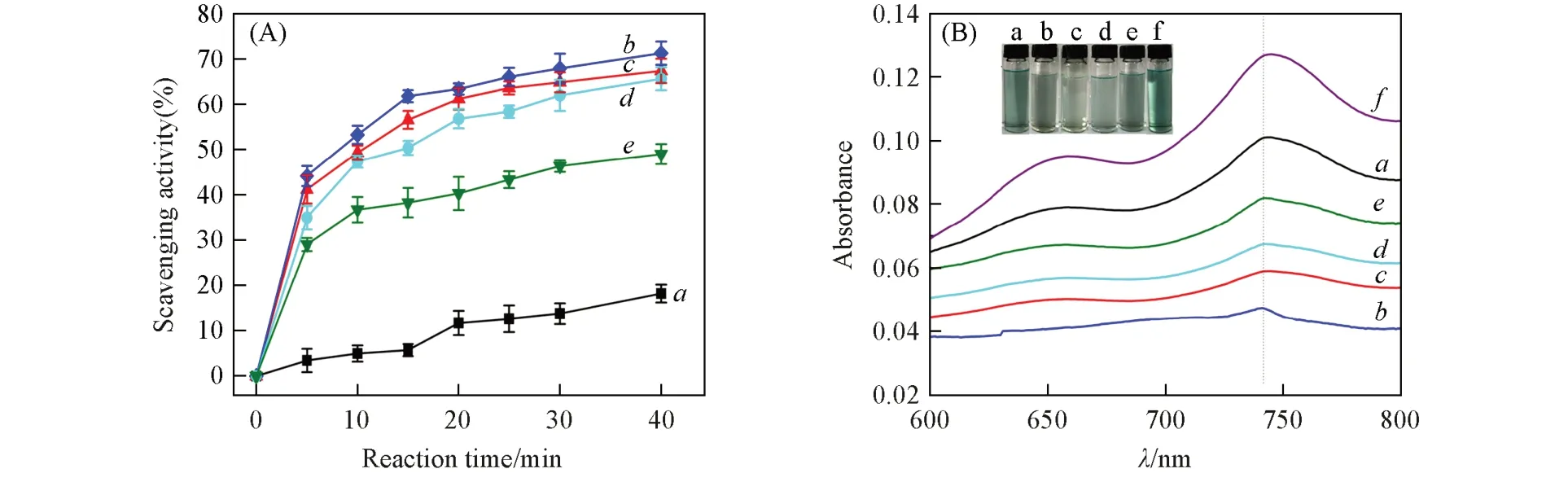
Fig.8 ABTS·+ radical scavenging activity(A) and UV absorption spectra(B) of ABTS solution after 40 min reaction with CTS(a), O-HACC-0.2-1/3-80-3(b), O-HAOCC-4-1(c), O-HAOCC-4-3(d)and O-HAOCC-4-6(e)(B) f. control. The inset shows the color change of ABTS·+ solution(f) after 40 min reaction with CTS(a),O-HACC-0.2-1/3-80-3(b),O-HAOCC-4-1(c),O-HAOCC-4-3(d) and O-HAOCC-4-6(e).
CTS,O-HACC 和O-HAOCC 中均存在伯氨基,具有较强的供电子能力,ABTS·+自由基能将氨基氧化成—C=NH,而O-HACC 和O-HAOCC 两种壳聚糖衍生物中都有季铵盐结构,其分子聚阳离子性比CTS增强,自由基清除率明显高于CTS[27],两种壳聚糖衍生物的抗氧化活性提高间接地表明壳聚糖在碱性条件下接枝上季铵盐基团. 另外,KIO4短时间氧化(≤3 h)只是将O-HACC分子中小部分的C2-NH2和C3-OH氧化为醛基,不会对O-HAOCC的抗氧化能力造成明显影响,但长时间氧化会使O-HACC中的羟基和氨基数目大量减少,导致其自由基清除率下降较大,这也进一步证明了经KIO4氧化后,季铵盐壳聚糖中生成了醛基.
2.4 O-HAOCC改性棉织物的制备与表征
图9 示出了不同质量浓度O-HAOCC 处理的改性棉织物的含氮量和接枝率. 由图9 可见,随着O-HAOCC浓度的提高,改性棉织物的含氮量和接枝率逐渐增加,当O-HAOCC的质量分数为2%时,改性棉织物的含氮量与接枝率分别达到0.84%和9.84%,而后随O-HAOCC 浓度进一步增大,改性织物的接枝率及含氮量增加缓慢,这表明棉织物上的反应位点有限,随着O-HAOCC添加量增多,接枝反应趋于饱和. 为了分析O-HAOCC与棉织物形成化学键接枝情况,对改性棉织物进行红外光谱测试,结果如图10 所示. 与原棉织物相比,接枝O-HAOCC 后的棉织物在1720.8 和2852.5 cm-1附近分别出现了O-HAOCC 中醛基C=O 和C—H 的伸缩振动峰,在1525.4 cm-1处出现O-HAOCC 的N—H 弯曲振动峰,且在1461.6 cm-1处出现季铵盐基团的C—H弯曲振动峰,说明O-HAOCC 成功接枝到棉织物上; 同时,随着O-HAOCC接枝率的增加,改性棉织物在893.4 cm-1附近的半缩醛吸收峰强度增大,进一步表明棉织物中的羟基与O-HAOCC分子中的醛基发生半缩醛反应[19,26],O-HAOCC通过半缩醛化学键与棉织物交联结合.

Fig.9 Effect of O-HAOCC mass concentration(w, %)on graft ratio(a) and nitrogen content(b) of modified cotton fabric
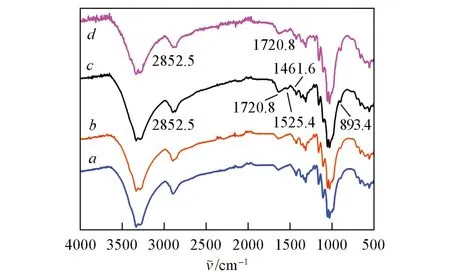
Fig.10 FTIR spectra of cotton fabric(a) and O-HAOCC modified cotton fabric with graft ratio of 4.81%(b), 9.84%(c) and 10.95%(d)
图11是原棉和不同接枝率O-HAOCC改性棉织物的SEM照片. 由图11可知,原棉纤维存在天然扭曲,表面较粗糙. 经过O-HAOCC接枝处理后,棉纤维呈现出不同形貌,表面附着有片状物,在接枝率达到9.84%时,棉纤维表面形成均匀薄膜,且有部分纤维粘合在在一起[图11(C)]; 但随着接枝率继续增加,O-HAOCC 会在织物表面形成大面积的交联膜,可能影响织物的使用舒适性[图11(D)]. 同时,通过EDS能谱分析可知,原棉织物中只含有C和O元素,而O-HAOCC改性棉织物上除了C和O元素外,还存在N和Cl两种元素,且两种元素含量随O-HAOCC接枝率提高而增加,说明O-HAOCC成功接枝到棉织物上.

Fig.11 SEM images(A1—D1, A2—D2) and EDS analysis(E—H) of cotton fabric(A1, A2, E) and O-HAOCC modified cotton fabric with graft ratio of 4.81%(B1, B2, F), 9.84%(C1, C2, G) and 10.95%(D1, D2, H)
2.5 改性棉织物的服用性能
表2为不同接枝率改性棉织物的服用性能测试数据. 从表2可以看出,O-HAOCC改性棉织物的断裂强力呈现增加趋势,可能是O-HAOCC 分子与棉纤维发生接枝反应,使改性棉纤维分子间作用力增强所致. 改性棉织物的毛细效应随着接枝率的增大而提高,在接枝率为9.84%时上升了36.63%,这是因为表面接枝O-HAOCC 的棉织物中含有更多的季铵盐和氨基、 醛基等亲水性基团,对水分子的吸附能力显著提升. 而O-HAOCC 改性棉织物的白度随接枝率增加而有较大下降,且实验中发现改性棉织物的颜色从白色变成淡黄色,这是由于O-HAOCC 本身颜色是淡黄色,用其改性导致改性棉织物白度降低. 经O-HAOCC 处理后的棉织物折皱回复角明显增大,接枝率9.84%的改性棉织物折皱回复角提高了43.06°,说明O-HAOCC 与棉纤维形成半缩醛交联,可在一定程度上增加改性棉织物的抗折皱性能.
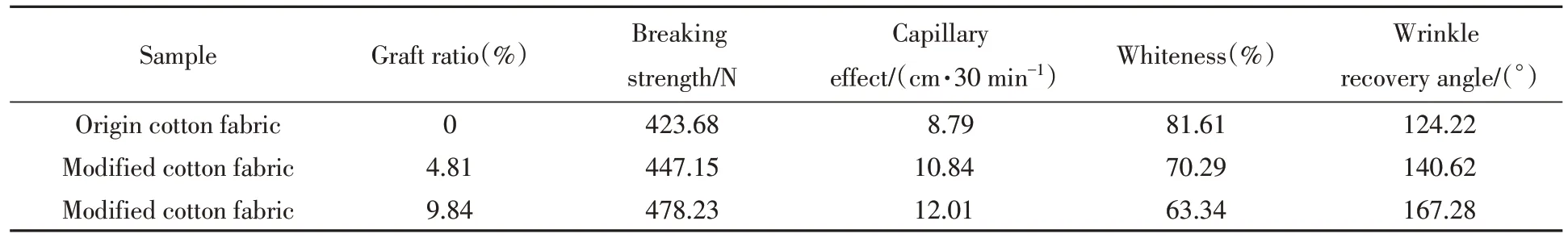
Table 2 Changes in wearability of O-HAOCC modified cotton fabric
表3为本文制备的改性棉织物与文献[19,20]中样品的服用性能对比数据. 从表3可以看出,实验制备的改性棉织物拥有更优良的服用性能,其断裂强力、 折皱回复角和毛细效应提升更显著,在实际使用中改性棉织物舒适度及耐用性更好.
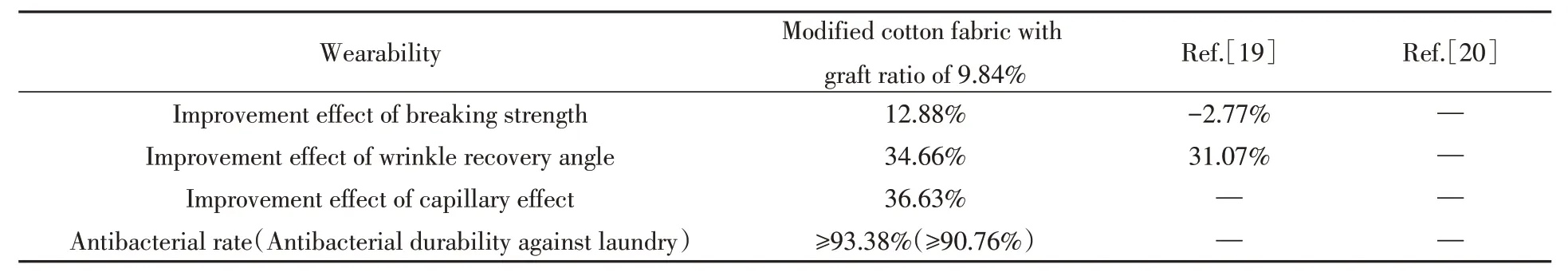
Table 3 The wearability of different materials
2.6 改性棉织物的抗菌性能
为了考察O-HAOCC改性棉织物的抗菌活性,采用美国AATCC Test Method 100-1999方法对金黄色葡萄球菌和大肠杆菌两种细菌进行定量抗菌测试,表4示出不同接枝率的改性棉织物的抗菌率数据.由表4可知,原棉织物上繁殖了大量细菌菌落,对S. aureus和E. coli几乎没有抗菌作用. 而O-HAOCC改性棉织物具有较强的抗菌效果,其中接枝率9.84%的改性棉织物对金黄色葡萄球菌和大肠杆菌的抗菌率分别高达98.63%与93.38%,且水洗50次后的改性棉织物仍保持了较高的了抗菌率(≥90.76%)达到FZ/T 73023-2006《抗菌针织品》的3A 级抗菌标准,表现出优异的抗菌耐洗涤性能. 这是因为O-HAOCC分子中含有聚阳离子氨基与季铵盐双重抗菌基团,同时O-HAOCC借助半缩醛化学键牢固接枝在棉织物表面,从而使改性棉织物拥有更强的持久抗菌能力.
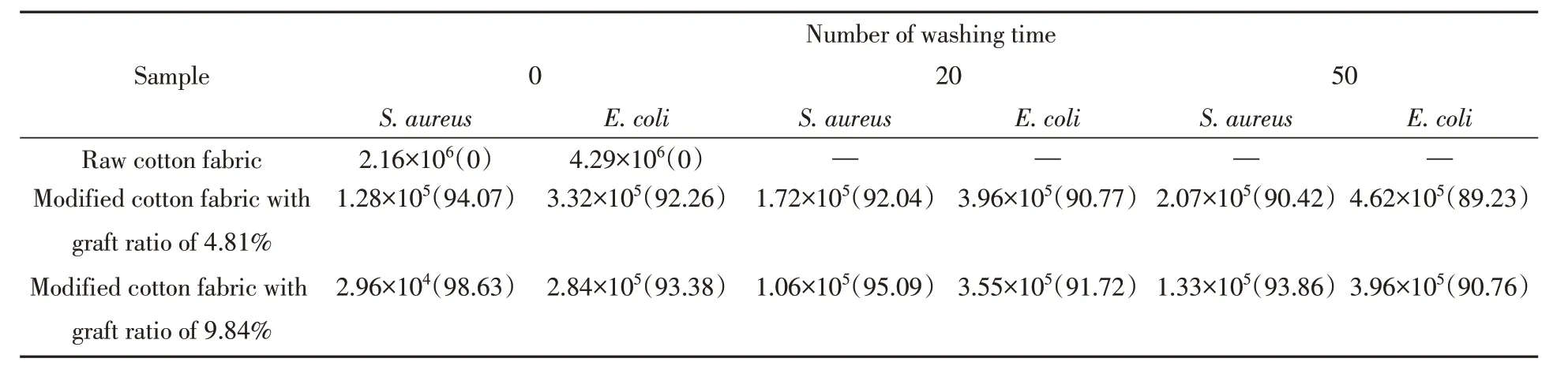
Table 4 Survival colony count(CFU/mL) and antibacterial rate(%,in parentheses) of cotton fabric before and after O-HAOCC modification
3 结 论
通过对壳聚糖的C2 位用席夫碱保护,并在其C6 位接枝季铵盐制备出O-HACC,然后利用KIO4局部选择性氧化O-HACC 的C2 和C3 位基团得到O-HAOCC.O-HAOCC 保持了壳聚糖的氨基和天然抗菌性,具有季铵盐和阳离子氨基的双重抗菌活性,同时生成了可与纤维化学键合的活性醛基. 季铵盐氧化壳聚糖的优化制备工艺为: Schiff-CTS 与GTA 摩尔比为1∶3,反应温度为80 ℃,NaOH 质量分数为0.2%,反应时间为3 h,KIO4浓度为4 g/L,氧化时间为3 h. FTIR,XRD,1H NMR,SEM和DSC等分析显示,季铵盐基团成功接枝在壳聚糖C6 位,且O-HACC 的部分C2 和C3 位形成醛基,O-HACC 与O-HAOCC 内部结构变得松散,热稳定性降低. 4 g/L KIO4氧化6 h的O-HAOCC 水溶性达到279 g/L,拓展了应用范围; 合成的O-HACC 和O-HAOCC 的均含有季铵盐结构和氨基,其自由基清除率分别为71.21%和65.58%,抗氧化能力较强. 当O-HAOCC 质量分数为2%、 反应时间为2 h 和接枝温度为80 ℃时,改性棉织物中的O-HAOCC 接枝率达到9.84%,O-HAOCC 与棉织物形成半缩醛化学键结合,O-HAOCC改性棉织物对金黄色葡萄球菌和大肠杆菌的抗菌率分别为98.63%和93.38%,水洗50次后的抗菌率仍在90.75%以上,可作为持久抗菌纺织材料而广泛应用.
猜你喜欢
纺织科技进展(2016年3期)2016-11-29
广州大学学报(自然科学版)(2015年4期)2015-12-23
纺织科技进展(2015年1期)2015-11-28
邵阳学院学报(自然科学版)(2015年2期)2015-06-05
丝绸(2015年11期)2015-02-28
现代纺织技术(2015年6期)2015-02-28
西安工程大学学报(2014年2期)2014-02-28
无机化学学报(2014年7期)2014-02-28
无机化学学报(2014年7期)2014-02-28
无机化学学报(2014年4期)2014-02-28
