
肝细胞内Ca2+信号与肝脏再生和肝脏疾病的联系
2024-03-25 04:33郝永乐邹仁哲韩静雯唐建红
赣南医学院学报 2024年1期
郝永乐,邹仁哲,韩静雯,陈 菲,唐建红
(1. 赣南医科大学基础医学院;2. 赣南医科大学第一临床医学院;3. 赣南医科大学康复学院;4. 赣南医科大学赣州市实验动物工程技术研究中心,江西 赣州 341000)
据统计,中国超过1/5 人口患有肝脏疾病,包括乙型肝炎病毒(Hepatitis B virus,HBV)和丙型肝炎病毒(Hepatitis C virus,HCV)感染、肝硬化、肝癌、非酒精性脂肪性肝病(Nonalcoholic fatty liver disease,NAFLD)和酒精相关性肝病等[1]。有研究[2]报道,全世界每年约有200多万人死于肝脏疾病(肝硬化、病毒性肝炎和肝癌),占全世界死亡人数的4%,其中有60 万~90 万人死于肝癌。肝脏疾病主要的致病因素有病毒感染、脂肪积聚、胆汁淤积、药物诱导和酒精滥用等。Ca2+在肝脏疾病中扮演着重要角色,参与多种肝脏生物学功能调节过程,包括解毒、营养代谢、胆汁分泌、肝内胆汁酸合成和肝脏再生等。正常的Ca2+调节对于维持肝细胞的正常功能和代谢至关重要。
Ca2+作为第二信使,在维持机体细胞稳态中起重要作用,参与调控肌肉收缩、神经元信号传递、激素分泌、受精和细胞生长等诸多细胞功能。细胞内Ca2+主要储存在内质网,内质网也被称为Ca2+库,其Ca2+水平高于细胞质。静息条件下,细胞外环境Ca2+水平是细胞质Ca2+水平的104倍[3]。细胞接收Ca2+信号主要来源于细胞外Ca2+内流和储存在内质网或肌浆网的Ca2+流动。Ca2+进入细胞的方式根据其调节机制主要分为电压门控通道(Voltage-gated calcium channel,VGCC)、受体门控通道(Receptor-operated channels,ROC)和Ca2+库控制的Ca2+通道(Store-operated channels,SOC)。这3个通道具有不同的动力学特性,VGCC 和ROC 能在短时间内产生大量的Ca2+内流,而SOC 允许持续的低浓度Ca2+内流。SOC 在Ca2+浓度低水平时激活,在Ca2+浓度高水平时抑制。
Ca2+在肝脏疾病中发挥重要作用,Ca2+信号失调是急性、慢性肝病的标志。研究表明,细胞质Ca2+浓度增加会激活Ca2+依赖性转录因子和激酶,使肝细胞加速进入细胞周期,促进肝脏再生[4]。因此,研究不同肝脏疾病中的Ca2+信号改变有助于深入了解肝脏疾病的发病机制和病程发展,为肝脏疾病的治疗提供可能的新方向。
1 Ca2+信号简述
Ca2+信号系统由广泛的分子信号构成,这些分子信号协同作用后驱动具有独特空间和时间特性的Ca2+信号。细胞膜内外以及细胞质和细胞器之间的Ca2+浓度梯度的维持及动态调控主要依赖于位于细胞膜、内质网以及线粒体上的Ca2+通道的改变。
Ca2+可以通过VGCC、ROC 和SOC 方式进入细胞,通过细胞膜Ca2+转运ATP 酶(Plasma membrane Ca2+-ATPase,PMCA)和Na+/Ca2+交换通道(Na+/Ca2+exchanger,NCX)从细胞质转运到细胞外。Ca2+从细胞外进入细胞质的关键途径之一是VGCC。VGCC在可兴奋细胞发生动作电位时被激活,相关膜蛋白响应膜去极化而打开,并允许Ca2+沿其电化学梯度流入。它们将Ca2+传导到细胞中,启动许多生理过程,包括肌肉收缩、神经递质分泌和基因转录等[5]。VGCC 有多种类型,根据其电压敏感性可分为高压激活通道和低压激活通道。高压激活Ca2+通道家族包括L 型(Cav1.1~Cav1.4)、P/Q 型(Cav2.1)、N 型(Cav2.2)和R 型(Cav2.3)通道,低压激活通道包括3 种不同类型的T 型(Cav3.1~Cav3.3)Ca2+通道[6]。这些Ca2+通道分布在不同细胞的细胞膜上,并完成特定的生理功能。例如,Cav1.1 通道仅在骨骼肌中表达,它们对肌肉收缩至关重要,而Cav2.2 通道仅存在于神经元中,它们触发神经递质的释放[6]。ROC 主要有N-甲基-D-天冬氨酸受体(N-methyl-Daspartatereceptor,NMDAR)、烟碱型乙酰胆碱受体(Nicotinic acetylcholine receptors,nAChR)和经典瞬时受体电位通道(Transient receptor potential canonical,TRPC)[7]。谷氨酸作为配体可以激活NMDAR,NMDAR 通常存在于中枢神经系统中的神经元突触后膜上,参与学习和记忆等功能[8]。nAChR 属于乙酰胆碱受体,通常也称离子型乙酰胆碱受体。这是一种非选择性的阳离子通道,允许Ca2+、Na+和K+通过。nAChR 的激活可以介导3 种类型的细胞质Ca2+信号:⑴Ca2+直接通过nAChR 内流;⑵通过nAChR介导的去极化激活的电压依赖性钙通道间接引起Ca2+内流;⑶通过内质网兰尼碱受体(Ryanodine receptor,RyR)和肌醇(1,4,5)-三磷酸受体(Inositol 1,4,5-trisphosphate receptor,IP3Rs)引起的钙诱导钙释放(由前两种来源触发)[9]。TRPC 中TRPC3 和TRPC6 通道是Ca2+渗透的非选择性阳离子通道,参与许多生理过程[10]。通过细胞膜排出细胞内Ca2+的主要方式有PMCA 和NCX。PMCA 也被称为质膜ATP 酶,是许多膜Ca2+运输蛋白之一,对维持细胞内游离Ca2+的低浓度及其信号功能至关重要。细胞内Ca2+浓度升高时,PMCA 通过天冬氨酸磷酸化为Ca2+逆浓度梯度泵出细胞膜提供能量[11]。同样,NCX 也是排出Ca2+主要通道,在生理条件下,NCX 利用电化学梯度介导3 个Na+进入细胞,1 个Ca2+排出细胞。在病理环境中,NCX 的反向模式占主导地位(即介导3 个Na+排出细胞,1 个Ca2+进入细胞),可显著改变细胞内Ca2+稳态,在细胞或系统水平上导致许多Ca2+依赖事件发生[12]。细胞Ca2+信号几乎调节机体生理的各个方面,每一个Ca2+信号的传导都有一个复杂的传导链,包括一系列感知细胞外信号的受体和Ca2+可渗透通道,这些细胞膜上的通道将Ca2+输送到细胞内或从细胞内存储释放Ca2+,形成特定的Ca2+信号。
Ca2+信号传入细胞后可以被内质网ATP 酶(Sarcoplasmic reticulum Ca2+-ATPase,SERCA)识别并将Ca2+泵入内质网腔。细胞受到刺激后,内质网上Ca2+通道蛋白(IP3R 和RyR)的Ca2+敏感性开始增强,排出过载的Ca2+[13]。内质网和线粒体是细胞内Ca2+储存最多的两个细胞器,内质网是细胞内最大的Ca2+库。正常情况下,内质网可以通过多种Ca2+泵和Ca2+通道防止Ca2+耗尽或过载。细胞受到刺激时,内质网Ca2+增加,IP3R 对IP3 更加敏感,同时也导致RyR受到刺激,内质网将过多的Ca2+释放到细胞质中,从内质网释放的Ca2+可以通过电压依赖性阴离子通道(Voltage-dependent anion-selective channel,VDAC)进入线粒体膜间隙[14]。
线粒体是细胞的能量站,也是细胞Ca2+信号传递枢纽。线粒体通过其外膜(Outer mitochondrial membrane,OMM)上的VDAC 和线粒体内膜(Inner mitochondrial membrane,IMM)上的线粒体钙单向转运体(Mitochondrial calcium uniporter,MCU)复合体摄取Ca2+,通过线粒体NCX(mNCX)排出Ca2+[13]。MCU复合体由两个部分组成:MCU通道形成亚基和调节蛋白。MCU 通道形成亚基由MCU、MCUb 和EMRE 构成,其主要负责Ca2+渗透[15]。调节蛋白主要包含MICU1、MICU2 和MICU3,这些调节蛋白在细胞质Ca2+水平升高时促进Ca2+进入线粒体[15]。MCU复合体激活的条件是周围Ca2+浓度高于细胞质峰值,位于内质网-线粒体接触点(也称为线粒体相关膜,Mitochondria-associated membranes,MAMs)处的MCU 复合体可被激活。MAMs 的主要功能是严格控制局部Ca2+浓度,Ca2+可以从内质网直接储存到线粒体,而不增加细胞质Ca2+浓度[16](图1)。
2 肝脏再生中的Ca2+信号传导
肝脏再生受到细胞因子、生长因子、激素和神经递质等的调控,它们与肝细胞相互作用,有助于部分肝切除后的肝脏质量和功能恢复。旁分泌、自分泌和内分泌相互作用推动肝细胞进入细胞周期,并在肝脏再生组织中维持正常的分化功能。在调控信号通路时,一些Ca2+动员激动剂包括去胰岛素、精氨酸加压素、ATP、表皮生长因子和肝细胞生长因子有助于肝脏再生[17]。
胰岛素通过影响细胞核Ca2+参与调控肝细胞增殖。胰岛素除了具有显著降血糖作用外,还具有促进有丝分裂作用,可以促进细胞增殖。胰岛素诱导分离的大鼠肝细胞发生Ca2+振荡,这些Ca2+信号依赖于磷脂酶C(Phospholipase C,PLC)和IP3R 的激活[18]。胰岛素通过胰岛素受体起作用,胰岛素受体是一种异四聚体受体酪氨酸激酶,由2 个具有配体结合活性的细胞外α 亚基和2 个具有酪氨酸激酶活性的跨膜β 亚基组成,在胰岛素的刺激下胰岛素受体由细胞膜转移到核内[19]。胰岛素与胰岛素受体结合后蛋白酪氨酸激酶被激活,β 亚基内酪氨酸残基发生磷酸化,导致磷脂酰肌醇3-激酶和PLC 配体蛋白的募集[20]。PLC 水解磷脂酰肌醇4,5-二磷酸,形成二酰基甘油(Diacyl glycerol,DAG),激活蛋白激酶C(Protein kinase C,PKC)及IP3,促进Ca2+从细胞内Ca2+库中释放结合到细胞核上的IP3R 上,可能是胰岛素对肝脏再生产生影响的作用机制[17]。
线粒体Ca2+通过抑制细胞凋亡调节肝细胞增殖,线粒体Ca2+通过调节Bax/Bcl-2 的比例,使肝细胞朝着抗凋亡的方向发展,从而加速肝切除术后肝脏恢复[21]。线粒体Ca2+还可以通过调节线粒体基质中ATP 的产生加速肝脏再生。此外,肝细胞生长因子是肝脏再生的重要刺激因子,在损伤组织中具有抗凋亡活性,并在细胞周期早期诱导细胞内Ca2+升高[22]。在肝细胞中,Bcl-2与凋亡受体结合后激活线粒体凋亡途径[23]。BH3 相互作用域凋亡激动剂(Bid)的缺失导致内质网储存Ca2+和细胞内Ca2+水平降低,影响了细胞周期蛋白D1和细胞周期蛋白E的表达,进而调节细胞周期进程[23]。此外,有研究[24]发现,MICU1敲除小鼠会出现线粒体Ca2+过载,进而诱导线粒体渗透性过渡孔(Mitochondrial permeability transition pore,mPTP)加速开放,导致肝脏再生失败,细胞周期受损和组织损伤可以通过抑制mPTP来预防。综上,线粒体Ca2+可以通过多种方式促进肝脏再生(图2)。

图2 胰岛素通过影响细胞核钙离子参与肝细胞再生
3 肝脏疾病中的Ca2+信号传导
许多肝脏功能通过细胞内Ca2+调控,如能量代谢、胆汁分泌、线粒体生理、细胞再生和基因表达等[25]。因此,Ca2+信号的异常转导也预示着肝细胞功能异常甚至肝脏疾病的发生(图3)。
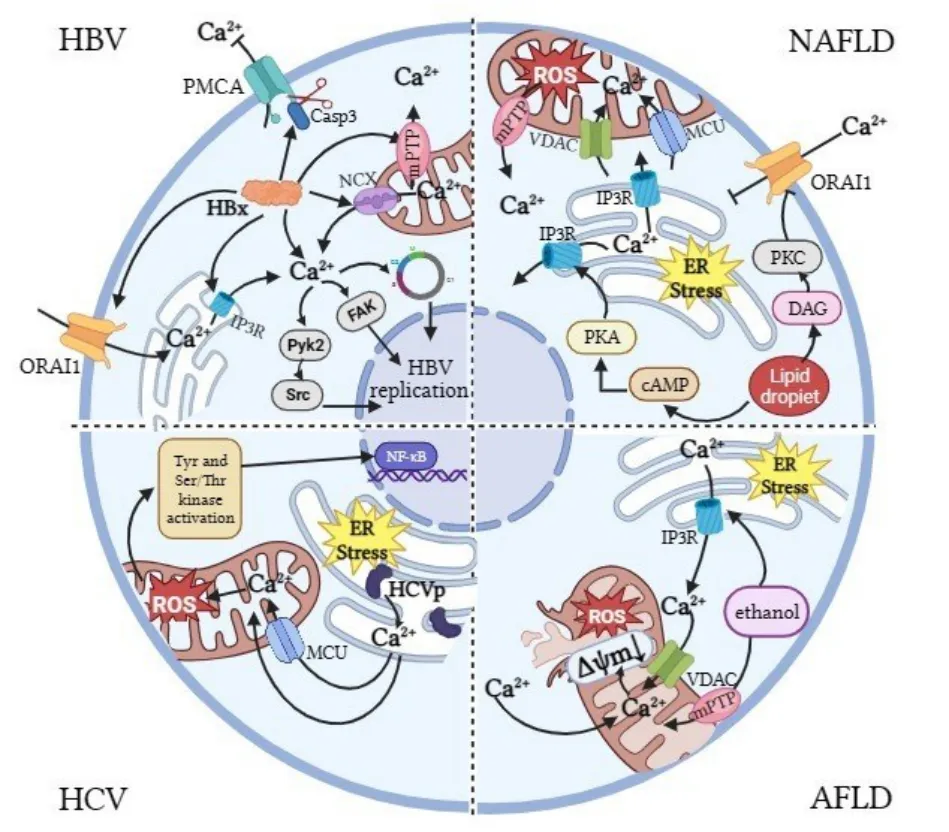
图3 不同肝脏疾病中钙离子信号紊乱示意图
3.1 慢性病毒性肝炎 HBV 和HCV 中的Ca2+信号转导及相关蛋白是近年来的研究热点。HBV 和HCV 感染肝脏细胞后引起细胞膜通透性增加和储存Ca2+的细胞器(如内质网和线粒体)膜通透性改变,导致细胞质Ca2+增加[26]。HBV 可以利用多种机制提高细胞质Ca2+水平,细胞质Ca2+的增加又能够促进病毒复制[27]。HBV 介导的Ca2+升高主要依赖于HBV 编码蛋白HBx。一方面,HBx 通过调控细胞膜、内质网和线粒体中包括钙释放激活Ca2+通道调节分子1、IP3R 和mPTP 在内的多个Ca2+通道的激活和表达,提高细胞质中Ca2+的水平[27]。另一方面,HBx 通过Ca2+信号依赖性的富含脯氨酸的酪氨酸激酶2/非受体酪氨酸激酶(Pyk2/Src)和黏着斑激酶途径激活来促进HBV 复制[27]。CHAMI M 等[28]发现,HBx 能够通过激活胱天蛋白酶3(Caspase-3)来抑制细胞膜上PMCA 的活性以避免Ca2+外流。Ca2+信号在病毒感染中起着重要作用,越来越多的证据表明,通过合适的药物控制细胞内Ca2+或Ca2+通道是控制HBV复制的潜在策略[29]。
HCV 主要由3 个结构蛋白(核心蛋白、E1 蛋白和E2 蛋白)和7 个非结构蛋白(p7 蛋白、NS2、NS3、NS4A、NS4B、NS5A 和NS5B)构成。病毒NS3-4A 蛋白酶裂解线粒体抗病毒信号蛋白并使其与线粒体膜分离,这种作用有助于病毒从先天免疫反应中逃逸[30]。尽管HCV 蛋白合成、组装折叠大多发生在内质网,但是核心蛋白和NS3-4A 主要定位于OMM。HCV 核心蛋白与线粒体功能密切相关,并通过增加活性氧(Reactive oxygen species,ROS)和活性氮的产生导致肝细胞氧化应激[31]。HORNER S M 等[32]认为HCV 蛋白在MAMs 中积聚。SCHWER B 等[33]提出HCV 蛋白通过MAMs 中部分膜的短暂融合从内质网横向扩散到线粒体。并且,HCV 复制会破坏正常内质网功能,诱发内质网应激,同时促进线粒体通过MCU 吸收Ca2+,导致ROS 升高。ROS 在细胞中累积过量时又反过来促进HCV 复制、mPTP 开放和肝细胞氧化应激[34]。
3.2 非酒精性脂肪性肝病 肝细胞内Ca2+浓度异常和细胞内慢性内质网应激是NAFLD 向非酒精性脂肪肝炎、胰岛素抵抗、肝硬化、肝纤维化和肝细胞癌发展的核心[35]。肝细胞内发生脂肪变性后细胞内Ca2+稳态显著改变。一方面,脂滴中存在的部分DAG 在细胞膜激活PKC 的异构体,即PKCε、PKCδ或PKCβ[36]。PKC 通过磷酸化OARI1(SOC 通道蛋白)抑制Ca2+进入内质网,内质网Ca2+浓度降低后导致内质网应激;另一方面,由于Ca2+转运蛋白的表达减少及内质网膜中磷脂酰胆碱比磷脂酰乙醇胺的比例升高对内质网ATP 酶2b(SERCA2b)活性的抑制导致内质网Ca2+摄取减少[36]。肝细胞表达3 种IP3R,即IP3R1、IP3R2 和IP3R3。IP3R1 主要参与MAMs 上Ca2+转移和内质网部分Ca2+释放,IP3R2 是肝细胞中主要的Ca2+通道,参与细胞质Ca2+调节,IP3R3 具有独特的Ca2+结合特性,在细胞质Ca2+浓度增加时保持开放状态[37]。有研究[38-39]表明,肝细胞脂肪变性后,cAMP 和蛋白激酶A(PKA)介导的磷酸化激活IP3R1,导致内质网Ca2+部分释放,IP3R2 受到损伤进而影响Ca2+信号的正常传导。肝细胞内脂质积累通过多种途径导致内质网Ca2+降低和线粒体及细胞质Ca2+升高。因此,抑制Ca2+从内质网传导到线粒体这个途径有助于治疗NAFLD。
3.3 酒精性脂肪肝病 酒精性脂肪肝病(Alcoholic fatty liver disease,AFLD)是由长期和过量饮酒引起的,是酒精性肝病最常见和可逆的阶段。长期和过量饮酒所导致的肝损伤,早期病理改变发生在线粒体中[40]。慢性乙醇摄入会导致线粒体功能障碍,包括mtDNA 和核糖体损伤、氧化磷酸化抑制和ROS生成增加[40]。游离Ca2+是调节线粒体内部功能的关键因素,并在多个水平上控制ATP 合成和ROS 产生。KING A L 等[41]发现,长期喂食乙醇的大鼠更容易发生肝脏线粒体Ca2+超负荷,线粒体Ca2+超负荷触发mPTP 的开放,随后可能增加ROS 的产生。其途径可能是Ca2+从细胞质转移到线粒体,增加线粒体膜通透性,降低线粒体膜电位,最终导致肝细胞损伤[42]。因此,维持细胞内Ca2+的平衡对于预防和治疗AFLD 非常重要。减少酒精摄入和加强对Ca2+的调节可以减少脂肪聚集和肝细胞损伤,从而降低AFLD的发病风险和病程。
3.4 胆汁淤积 肝细胞的主要生理功能之一是分泌胆汁,各种转运系统抑制、细胞骨架中毒、细胞内Ca2+稳态紊乱和胆汁成分反流等多种机制会引起肝细胞内胆汁淤积的发生。胆汁淤积可引起肝纤维化、胆汁性肝硬化和终末期肝病。在胆汁淤积性疾病的发病过程中Ca2+信号通路起着重要作用[43]。胆汁酸是胆汁的主要溶质,其在胆总管胆汁流量中起重要作用。胆汁酸对Ca2+有高亲和力,可以与其形成胆汁酸-钙复合体。胆汁酸可通过促进细胞摄取Ca2+和增加细胞内游离Ca2+影响细胞代谢,从而引起胆汁淤积[44]。细胞内Ca2+动员激动剂可以诱导短期的胆汁流动效应。胰高血糖素本身不是Ca2+的激动剂,但它能够调节Ca2+动员激动剂(如抗利尿激素)对胆汁流动起作用。
IP3R 介导肝细胞和胆管细胞中Ca2+信号传导,IP3R介导的胆管上皮Ca2+信号通路对正常胆汁分泌很重要。胆汁淤积发生时,IP3Rs 在胆管上皮中损伤IP3介导的Ca2+信号通路,这可能与胆汁淤积的病理生理相关[45]。同时,胆汁淤积会损害细胞内Ca2+通道蛋白的表达,并导致胆道上皮细胞的信号传递受阻[45](图4)。因此,调节肝细胞和胆管细胞Ca2+信号通路可能为治疗胆汁淤积性疾病提供一种新的方法。
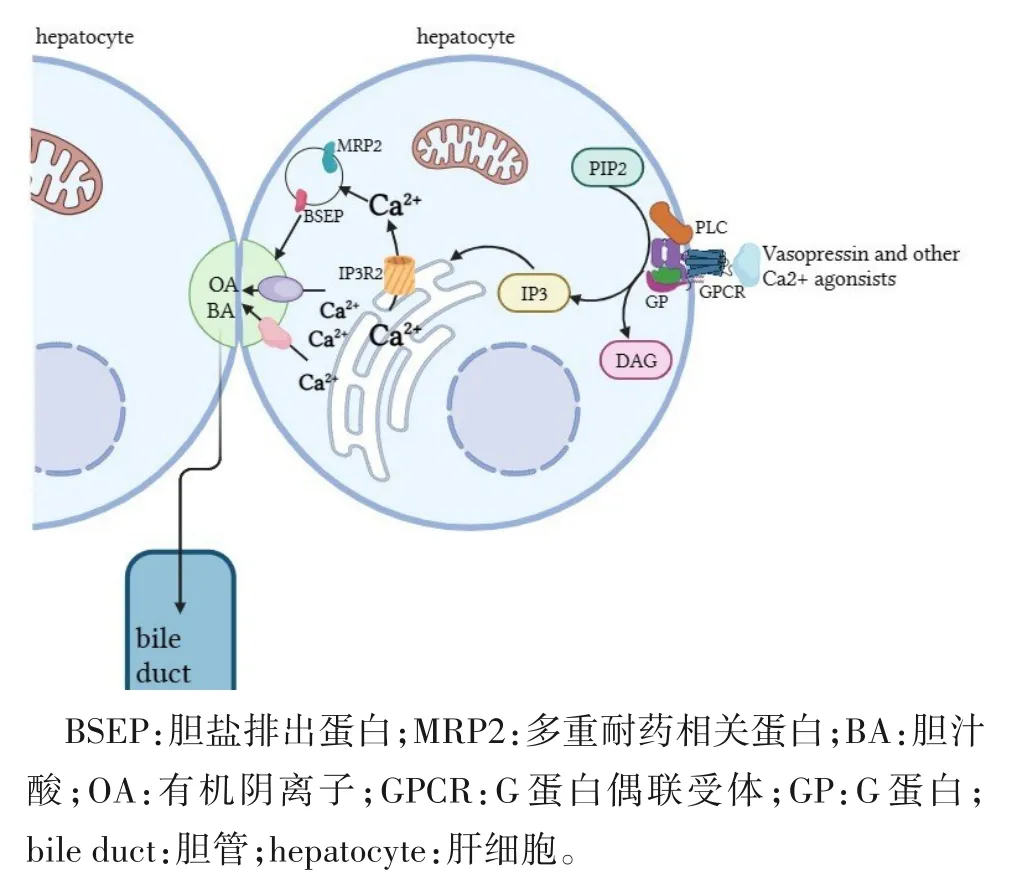
图4 IP3R介导的钙离子信号传导图
4 小结
Ca2+在肝脏再生和许多肝脏疾病发病过程中起着至关重要的作用。多种慢性肝病的特征是细胞质Ca2+信号的改变,其伴随着内质网Ca2+的消耗、细胞核Ca2+以及线粒体Ca2+的改变。病毒感染和脂质积累都会导致内质网应激,促进线粒体吸收Ca2+。胆汁淤积可由多种原因导致,胆管细胞中IP3R介导的Ca2+信号通路不仅是导管分泌的一种途径,并且对肝脏正常胆汁分泌非常重要。胆汁酸通过与Ca2+结合的方式促进细胞Ca2+摄取,导致游离Ca2+增加破坏细胞代谢,引起胆汁淤积。Ca2+影响肝脏再生则是通过多种途径,细胞质Ca2+促进肝细胞进入增殖周期,线粒体Ca2+通过抑制细胞凋亡促进肝细胞增殖。肝脏疾病发展过程中,炎症、氧化应激、脂肪积累等多种因素导致Ca2+信号传导通路紊乱,进而影响细胞的生理功能。因为Ca2+信号在肝脏疾病发病过程中的重要作用,Ca2+通道的改变未来可能作为肝脏再生或肝脏疾病的诊断标志物。Ca2+拮抗剂可以缓解部分肝脏疾病,如NAFLD、肝纤维化和肝硬化等,但Ca2+拮抗剂对正常肝脏、肝脏再生和肝脏疾病患者的影响还需要进一步研究。
猜你喜欢
科学导报(2024年20期)2024-04-22
解放军医学杂志(2021年12期)2022-01-18
现代临床医学(2021年1期)2021-01-26
肝博士(2020年5期)2021-01-18
肝博士(2020年5期)2021-01-18
安徽医科大学学报(2016年12期)2017-01-15
海南医学(2016年8期)2016-06-08
上海农业学报(2016年5期)2016-02-10
中国蔬菜(2015年9期)2015-12-21
应用海洋学学报(2015年3期)2015-11-22
