
注射用交联透明质酸钠凝胶生物相容性研究
2024-03-20 13:02:00王丽洁梁羽朱雨婷孙国伟苏州苏大卫生与环境技术研究所有限公司江苏苏州215123
中国医疗器械信息 2024年3期
王丽洁 梁羽 朱雨婷 孙国伟 苏州苏大卫生与环境技术研究所有限公司 (江苏 苏州 215123)
内容提要: 目的:评价注射用交联透明质酸钠凝胶的生物相容性。方法:分别通过细胞毒性试验、致敏试验、皮内反应试验、急性毒性试验、遗传毒性试验、植入试验、慢性毒性试验来全面评价注射用交联透明质酸钠凝胶的生物相容性。结果:试验结果显示,注射用交联透明质酸钠凝胶对L929细胞有轻度细胞毒性、无致敏反应、无遗传毒性、无急性和慢性毒性反应、皮内反应记分为1.5,植入试验4、13、26、52周各阶段评分为3.08、1.83、0.33、0,52周基本完成降解。结论:该注射用交联透明质酸钠凝胶对局部组织有轻微不良反应,但未发现全身毒性和遗传毒性影响。
透明质酸又名玻尿酸,为双糖(D-葡萄糖醛酸和D-N乙酰氨基葡萄糖)重复构成的直链结构,主要以钠盐的形式存在,在人体内主要分布在皮肤、结缔组织、关节腔滑液等组织中。由于其没有种属和组织特异性,被广泛用于临床如骨科润滑补充剂、术后防粘连剂、眼科黏弹剂及皮肤填充剂等产品[1]。其中用于皮肤填充的交联透明质酸钠凝胶,由于其在组织局部保留时间长,可通过注射实现微创填充美容,近年来受到医美界的关注[2,3]。但交联透明质酸钠凝胶由于其生产工艺中引入了添加剂和交联剂,其残留量的控制将直接影响生物学反应,交联反应导致的原材料分子结构变化和植入后体内酶解中间产物均有可能导致生物学反应[4,5]。本研究按照ISO 10993.1-2018[6]设计方案策略,并按ISO系列标准[7-11]
要求开展细胞毒性试验、致敏试验、皮内反应试验、急性毒性试验、遗传毒性试验、植入试验、慢性毒性试验进行生物学评价,为这一产品的后续的申报注册及临床应用奠定基础。
1.材料与方法
1.1 一般材料
研究起止时间:2020年1~2021年5月。
仪器设备:高压灭菌器;CO2培养箱;倒置显微镜;酶联免疫检测仪;电子天平;超净工作台;恒温振荡器;电子秤;恒温水浴锅;离心机;生物安全柜;生化培养箱;生物显微镜等。
试剂:注射用交联透明质酸钠凝胶(由某单位提供);胎牛血清(GiBco);1640培养基(HyClone);胰酶(GiBco);青霉素链霉素(GiBco);MEM(GiBco);PBS(GiBco);中性红(Sigma);弗氏完全佐剂(Sigma);十二烷基硫酸钠(国药集团);秋水仙素(国药集团);DMSO(国药集团);S9(MOLECULAR TOXICOLOGY,INC);甲磺酸甲酯(Sigma);环磷酰胺(RHAWN);丝裂霉素(GLPBIO);9-氨基吖啶(Sigma);2-硝基芴(Sigma);2-氨基蒽(Sigma);苯并(a)芘(Aladdin);叠氮钠(Alfa Aesar);吉姆萨染液(Solarbio);三氟胸苷(Sigma);L-组氨酸(国药集团);D-生物素(国药集团)。
试验系统:小鼠成纤维细胞(L929);鼠伤寒沙门氏菌(TA97a,TA98,TA100,TA102,TA1535);小鼠淋巴瘤细胞(L5178Ytk+/-);中国仓鼠肺细胞(CHL);新西兰白兔(苏州高新区镇湖实验动物科技有限公司);白化短毛豚鼠(上海甲干生物科技有限公司);ICR小鼠、SD大鼠(上海斯莱克实验动物有限责任公司)。
1.2 方法
1.2.1 细胞试验
按照ISO 10993-5:2009方法进行试验评价细胞毒性。以10mm×10mm滤纸为载体,涂布样品用于试验组。阳性组为天然乳胶,阴性组为高密度聚乙烯,每组分别设3个平行。将样品覆盖于L929细胞层,于37°C 5% CO2培养箱中培养48h,取出平皿在底部用标记笔标出样品所在位置,弃样品并去除培养基,在每皿中加入2mL中性红孵育1h,吸弃中性红,加入2mL PBS,显微镜观察细胞形态变化。
1.2.2 致敏试验
按照ISO 10993-10:2009方法进行试验,评价样品潜在的皮肤致敏反应。采用豚鼠最大限度试验,试验组以样品原液对豚鼠背部进行6点位皮内注射诱导,注射后7d以样品于皮内注射部位进行48h局部贴敷诱导,局部诱导后14d于侧腹部进行24h敷贴加以激发,除去样品后24、48h观察皮肤红斑和水肿并记分。
1.2.3 皮内反应试验
按照ISO 10993-10:2009方法进行试验,采用原液皮内注射方式给药。动物背部剃毛后在左侧指定区域注射5个位点,每点注射0.2mL样品原液,右侧区域注射对照生理盐水。注射后0、24、48、72h观察各注射局部和周围皮肤反应,包括红斑、水肿、坏死等,并按照标准记分方法进行记录。
1.2.4 急性毒性试验
按照ISO 10993-11:2017方法进行试验。取10只SD大鼠,雌雄各半,采用原液进行一次性腹腔注射给药,注射剂量15mL/kg体重(相当于体重70kg成人在100倍安全系数下注射10mL),对照组给予等剂量生理盐水。注射后观察小鼠即时反应,并于4、24、48和72h观察和记录动物的一般状态,毒性表现和死亡动物数。
1.2.5 慢性毒性试验
按照ISO 10993-11:2017方法进行试验。取60只SD大鼠,雌雄各半,以原液在动物背部进行多点皮下注射给药,注射总剂量15mL/kg体重,对照组植入高密度聚乙烯对照。试验周期180d。每天对动物进行临床观察,每周称重。试验终止日前一晚禁食过夜,终止日采血进行血液学和血液生化检查。对动物进行大体解剖观察,称取脏器湿重,将器官和组织保存于10%甲醛固定液中。用组织学方法处理组织(包埋、切片、苏木精-伊红染色),进行组织病理学检查。
1.2.6 鼠伤寒沙门氏菌回复突变试验
按照ISO 10993-3:2014和ISO/TR 10993-33:2015方法进行试验。试验组将样品按5µL/皿分别加入有、无活化系统S9的顶层培养基中,阴性对照和阳性对照分别用等体积的生理盐水和阳性诱变剂。于37°C培养72h,观察菌落生长情况,计数每皿回复突变的菌落数,用平均数和标准差来表示。
1.2.7 染色体畸变试验
按照ISO 10993-3:2014和ISO/TR 10993-33:2015方法进行试验。试验组将样品以5µL/mL(样品与试验系统终体积比)在有、无活化系统S9情况下,与中国仓鼠肺细胞共孵育4h和(或)24h。阴性对照和阳性对照分别加入等体积的生理盐水和阳性诱变剂。染毒结束后继续培养,加入秋水仙素。收集细胞,低渗及固定。滴片,吉姆萨染液染色,中性树胶封片及镜检阅片。每一剂量组至少选200个分散良好的中期分裂相进行染色体畸变分析,计算畸变率,用平均数和标准差来表示。
1.2.8 基因突变试验
按照ISO 10993-3:2014和ISO/TR 10993-33:2015方法进行试验。试验组将样品以5µL/mL(样品与试验系统终体积比)在有、无活化系统S9情况下,与L5178Y tk+/-细胞共孵育3h和(或)24h。阴性对照和阳性对照分别加入等体积的生理盐水和阳性诱变剂。在三氟胸苷选择下计数大、小集落数,计算抗性突变频率(Mutation Frequency,MF)和小集落突变百分比(Small Colony Mutation Frequency,SCMF)。
1.2.9 皮下植入试验
按照ISO 10993-6:2016方法进行试验。动物背部脊柱左侧皮下进行3个部位注射,每个部位0.2mL试验样品,右侧对照植入同类已上市产品,于4、13、26、52周4个观察期分别处死动物,观察植入部位情况并取材进行组织病理学检查。
2.结果
2.1 细胞试验结果
细胞毒性的大小用反应分级来表示,结果见表1。试验组细胞出现变性或裂解,反应局限于样品下区域,细胞毒性为2级轻度反应。

表1.细胞毒性反应(n=3)
2.2 致敏试验结果
试验组和阴性对照组动物激发后均未见皮肤红斑和水肿反应,致敏率为0%,表明样品在豚鼠未发现皮肤致敏反应。
2.3 皮内反应结果
皮内反应试验结果见表2。试验组皮内反应超过对照组,平均记分之差>1.0,不符合标准要求。继续观察7d,注射部位周围皮肤红斑消退,说明该损伤是可逆的。但试验样品仍存于皮内,形成隆起,延长观察至14d仍未有变化。

表2.皮内反应结果(n=3,分)
2.4 急性毒性试验结果
注射完毕后,试验组和对照组大鼠均未出现动物死亡,未见任何毒性反应,精神状态良好,体重增长在正常范围内。
2.5 慢性毒性试验
试验组及对照组大鼠均未观察到临床异常表现,两组大鼠试验前后的体重变化无显著差异(P>0.05)。动物大体解剖观察未见异常,试验组和对照组脏器系数亦无显著差异(P>0.05)。试验组个别血液学(MCH、MPV)和血液生化参数(ALB、P、K)与对照组相比有差异(P<0.05),但均在大鼠正常生物学范围之内,无实际生物学意义。试验组和对照组比较,未见明显组织病理学差异。
2.6 鼠伤寒沙门氏菌回复突变试验
试验背景菌苔生长良好,各菌株自发回复突变菌落数在预期数据范围内,在有、无活化系统情况下,阳性组各菌株突变数较阴性组均有显著增加(P<0.05)且在试验室历史数据范围内,试验组各菌株突变数与阴性组比较无明显差异(P>0.05),见表3。
表3.鼠伤寒沙门氏菌回复突变试验结果(n=3,±s,CFU/皿)
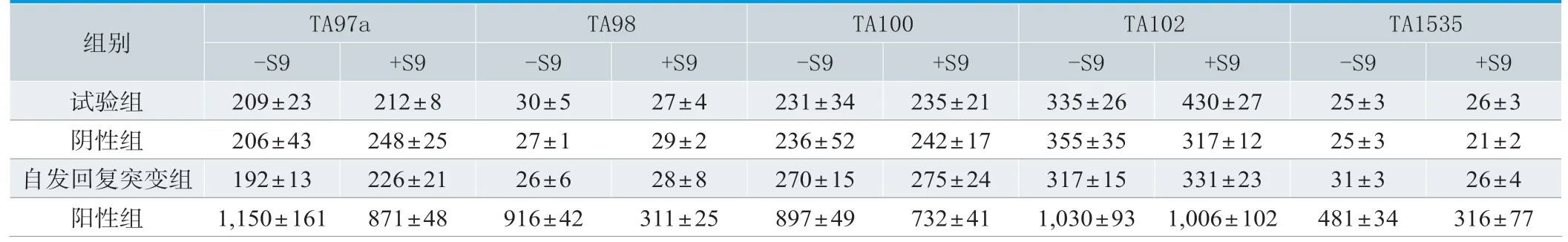
表3.鼠伤寒沙门氏菌回复突变试验结果(n=3,±s,CFU/皿)
组别TA97a TA98 TA100 TA102 TA1535-S9+S9-S9+S9-S9+S9-S9+S9-S9+S9试验组209±23 212±8 30±5 27±4 231±34 235±21 335±26 430±27 25±3 26±3阴性组206±43 248±25 27±1 29±2 236±52 242±17 355±35 317±12 25±3 21±2自发回复突变组192±13 226±21 26±6 28±8 270±15 275±24 317±15 331±23 31±3 26±4阳性组1,150±161 871±48 916±42 311±25 897±49 732±41 1,030±93 1,006±102 481±34 316±77
2.7 染色体畸变试验
在有、无活化系统情况下,经4h和(或)24h受试物暴露,试验组结构畸变率、细胞畸变率和阴性组比较均无明显差异(P>0.05),而阳性组结构畸变率和细胞畸变率较阴性组有显著差异(P<0.05),见表4。
表4.染色体畸变试验结果(n=3,±s,%)

表4.染色体畸变试验结果(n=3,±s,%)
组别-S9(4h)-S9(24h)+S9(4h)结构畸变率细胞畸变率结构畸变率细胞畸变率结构畸变率细胞畸变率试验组2.5±0.71 2.5±0.71 2.5±0.71 2.5±0.71 3.0±0.00 3.0±0.00阴性组1.5±2.12 1.5±2.12 2.0±1.41 2.0±1.41 2.0±0.00 2.0±0.00阳性组24.5±0.71 31.0±8.49 24.5±2.12 31.5±10.61 24.5±3.54 31.5±7.78
2.8 基因突变试验
在有、无活化系统情况下,经3h和(或)24h受试物暴露,试验组抗性MF及SCMF与阴性对照组比较无明显差异(P>0.05),阳性组MF和SCMF较阴性组有显著差异(P<0.05),见表5。
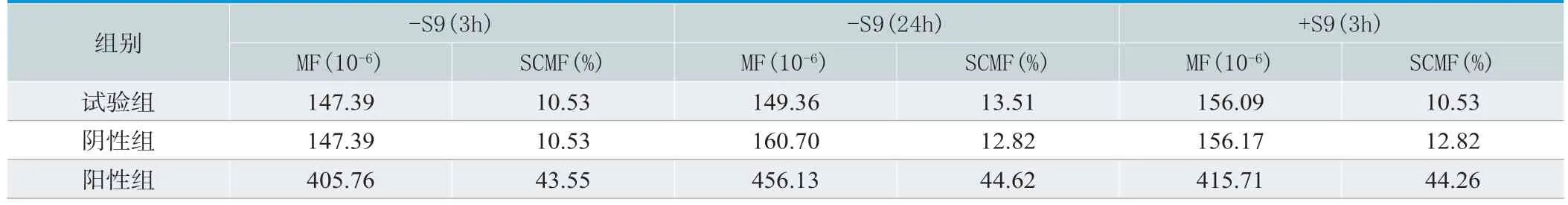
表5.基因突变试验结果(n=3)
2.9 皮下植入试验
植入后4、13、26、52周处死动物,分离皮下组织,肉眼观察4、13周植入物包囊明显,26周时植入物可见,52周时未见植入物包囊。植入后4、13、26周显微镜检查,可见不同程度纤维化包囊及炎症反应,包囊内可以观察到大量样品残留和碎片(见图1),组织病理学计分分别为3.08、1.83和0.33。植入52周后,病理观察基本无样品残留,炎症反应消失,记分为0。
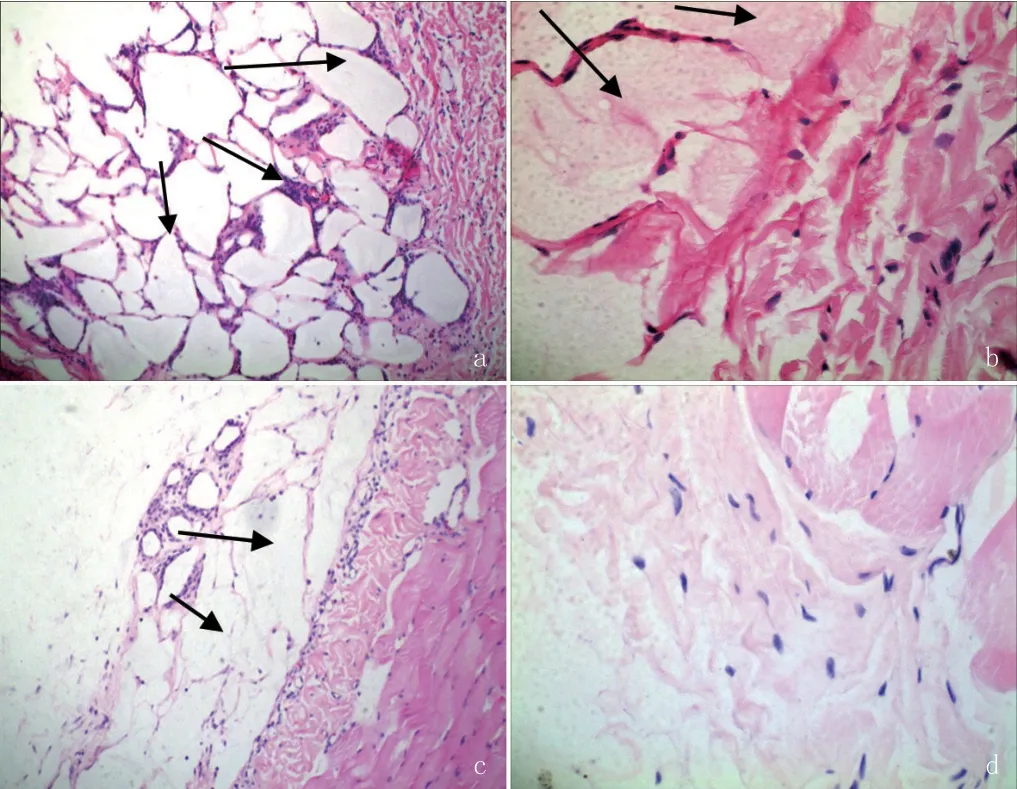
图1.皮下植入部位组织病理学观察(注:a.×100,4 周;b.×400,13 周;c.×100,26 周;d.×400,52 周;苏木精-伊红染色,黑色箭头所指为样品残留)
3.讨论
临床上透明质酸凝胶用于美容皮肤填充时,一般直接皮内或皮下注射至人体组织,故本研究体外试验中用样品原液直接和试验系统接触,体内动物试验时均模拟临床使用方式或采取更严格的接触途径开展试验,充分评估透明质酸凝胶生物安全性。
透明质酸进入人体后容易被代谢吸收,为维持填充的效果,用于医美时一般需加入交联剂及添加剂合成大分子结构,使其降解速度变慢。有文献指出,常用交联剂1,4-丁二醇二缩水甘油醚与细胞接触会引起一定毒性和炎症应[5]。本研究中细胞毒性、皮内反应、植入等局部试验中出现一定程度的毒性、刺激和炎症反应,可能是由交联剂的残留造成的,故生产商在产品生产合成工艺中应考虑交联剂的添加量和透明质酸的交联程度的合理控制,并对交联剂及其他添加剂残留进行有效去除,在保持合理降解速度维持填充效果的同时,尽量减少交联剂和添加剂对局部组织造成的不良反应。另外,由于透明质酸的可降解性,植入后按预估降解周期设定4个观察期,考虑覆盖降解开始、降解中期、降解完全整个过程,以充分研究产品降解吸收全过程对局部组织的影响[11]。结果表明,交联透明质酸凝胶在植入后6个月基本吸收完全,仅有部分切片可看到包囊残留。
在全身毒性试验中,急性毒性采用比皮下接触更严苛的接触途径腹腔注射,慢性毒性则模拟临床使用行皮下注射,且选择全身毒性暴露剂量为15mL/kg体重,为该产品人临床使用剂量(按70kg成人注射10mL计算)的100倍,充分考虑由动物外推至人体的可能性,评估该产品临床使用的风险。在遗传毒性试验中,由于样品为液体,且与试验系统相容,故采用极限暴露剂量5µL/皿或5µL/mL,另外在非活化系统下延长暴露期,从而最大程度考察样品的遗传毒性[12]。
根据ISO 10993-1要求,通过局部反应、全身毒性、遗传毒性试验等全面评估交联透明质酸钠凝胶的生物相容性,证明透明质酸钠凝胶具有比较好的生物相容性,但要注意控制和去除生产过程中可能引入的交联剂和其他添加剂残留,避免外源性物质对局部组织造成不良反应,以使最终成品满足临床安全性应用。后期还可以从交联残留定量检测或生产工艺控制和生物学试验间的反应联系等开展研究,为该产品的研发生产和临床应用奠定基础[13]。
猜你喜欢
云南化工(2021年11期)2022-01-12 06:06:08
纺织科学研究(2021年6期)2021-12-02 20:32:56
小哥白尼(野生动物)(2019年5期)2019-08-27 00:53:38
摄影之友(影像视觉)(2017年10期)2017-11-07 02:37:15
材料科学与工程学报(2016年4期)2017-01-15 13:35:49
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:10
中国医疗美容(2015年2期)2015-07-19 10:11:59
发明与创新(2015年33期)2015-02-27 10:40:02
癌变·畸变·突变(2014年2期)2014-03-01 04:39:41
养殖与饲料(2014年10期)2014-02-28 22:14:58
