
高效液相色谱法同时测定牙膏中六种氯酚类防腐剂
2024-03-18 04:38:22夏泽敏聂明霞黄敏涵李鑫宇谭建华席绍峰
口腔护理用品工业 2024年1期
廖 娜 夏泽敏 汪 毅 聂明霞 黄敏涵 李鑫宇 谭建华 席绍峰
(广州质量监督检测研究院,广州 511447)
氯酚类化合物(chlorophenols, CPs)是氯取代苯酚类化合物的总称,因具有良好的杀菌杀虫效果,被广泛用作杀菌剂、防腐剂[1],也被作为杀菌防腐剂广泛使用在口腔清洁护理产品中。然而,CPs是广泛的内分泌干扰物,对生物体具有致癌、致畸、致突变的“三致”效应。其性质稳定,难以生物降解,能在环境中相对持久地存在,且易通过食物链在生物体内富集[2],会对人体健康造成不利影响,因此多种CPs已被多国列入优先控制的毒性污染物名单。在我国,《牙膏用原料规范(GB 22115-2008)》中对几种氯酚类防腐剂有明确规定:双氯酚为牙膏中限用组分,牙膏中最大允许使用浓度为0.5%,并且需在产品标签中注明“含双氯酚”;溴氯芬、苄氯酚、氯二甲酚作为牙膏中许用防腐剂,其最大使用浓度分别为0.1%、0.2%、0.5%;六氯酚和p-氯-m-甲酚为牙膏中禁用组分。
通过对国内外相关标准和文献资料进行检索,近年来在个人护理产品中CPs的检测方面,检测方法主要有高效液相色谱法[3~6]、气相色谱法[7]和气相色谱-质谱联用法[8,],涉及的产品类型主要是化妆品、香皂等皮肤清洁用品。然而,化妆品和牙膏在配方方面存在较大差异,化妆品的方法标准仅可以作为参考,而在分析实际样品时,样品的溶解、均质、提取、净化、测定等操作均不能完全通用。因此,本方法拟采用高效液相色谱法测定牙膏中p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚、 溴氯芬、六氯酚等六种氯酚类防腐剂,为广大日化行业的检测机构和企业用户提供一种快捷简便、准确高效的测定方法。
1 实验部分
1.1 仪器与试剂
高效液相色谱仪(美国安捷伦公司);Milli-Q超纯水器(美国Millipore公司);IKA MS3 digital涡旋振荡器(德国IKA公司);SK8200H超声波清洗器(上海科导超声仪器有限公司);BS 224S电子天平(德国赛多利斯公司)。
p-氯-m-甲酚(CAS号35421-08-0)、六氯酚(CAS号70-30-4 )、双氯酚(CAS号97-23-4)、氯二甲酚(CAS号88-04-0)、苄氯酚(CAS号120-32-1)及溴氯芬(CAS号15435-29-7),纯度均大于98%,德国Dr.Ehrenstorfer公司;甲醇,色谱纯,德国Merck公司;乙腈,色谱纯,德国Merck公司;甲酸,分析纯,上海安谱科学仪器有限公司;石英砂,分析纯,广州化学试剂厂,超纯水,电阻率为18.2 MΩ·cm。
乙腈溶液(70%,v/v):准确移取700mL乙腈置于适量水中,再加水稀释至1000mL,混匀。
甲酸溶液(0.1%,v/v):准确移取1mL甲酸置于适量水中,再加水稀释至1000mL,混匀。
1.2 标准溶液的配制
分别准确称取p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚、溴氯芬及六氯酚的标准品10mg (精确至0.1mg)与10mL容量瓶中,用70%乙腈溶液定容至刻度,配制成质量浓度约为1000mg/L的标准储备液。使用时,稀释至所需要的质量浓度的标准工作溶液。
1.3 样品处理
将牙膏试样挤出约20mm后,准确称取试样1g(精确到0.001g)于50mL聚丙烯离心管中,加入约1g石英砂,涡旋后准确加入70%乙腈溶液20mL,涡旋混匀后超声提取10min,取适量提取液以10000r/min离心5min,上清液经0.22μm滤膜过滤后待测。
1.4 仪器条件
色谱条件:色谱柱:Weltch XB-C18(150mm×4.6mm,5μm);流动相:A:0.1甲酸,B:乙腈;梯度洗脱程序(0~2min,70%~50% A;2~7min,50% A;7~12min,50%~10% A;12~19min,10% A;19~19.1min,10%~70% A;19. 1~25min,70% A);流速:0.8mL/min;柱温:30℃;进样体积:20μL;检测波长:六氯酚、溴氯芬检测波长为298nm,p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚检测波长为282nm。
2 结果与讨论
2.1 提取方式及溶解的选择
根据p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚、溴氯芬及六氯酚等六种氯酚的结构以及理化性质,对提取溶剂和提取方法进行考察,筛选合理的提取方法。六种氯酚皆不溶或微溶于水,易溶于多数有机溶剂,考虑到牙膏产品在水中分散效果较好,本方法采用有机溶剂-水体系作为提取溶剂。考察了不同比例的甲醇水溶液和乙腈水溶液(0%、20%、40%、50%、60%、80%、100%)两种溶剂体系对含有六种氯酚的提取效果。结果显示,随着有机相比例增加,样液变澄清,样液变得容易经滤膜过滤,当有机相比例超过90%,样品容易成团,不易分散。在乙腈水溶剂体系中,不同比例有机溶剂提取的目标化合物回收率相似,皆能达到90%。在甲醇水体系中,随着甲醇比例增加,回收率逐渐增加,甲醇比例达到70%,回收率高于90%。综合两个溶剂体系对六种氯酚的提取情况来看,70%乙腈水溶液的响应面积最高。同时,考虑到牙膏一般具有较好的水分散性,提取溶剂中有机溶剂的比例越高,牙膏在提取溶剂中的分散效果越差。因此选择70%乙腈水作为六种氯酚的提取溶剂。另外,为了提高牙膏在提取溶剂中的分散效果,本方法选择加入适量石英砂对牙膏试样进行辅助分散。
2.2 色谱柱的选择
考察了Athena C8柱(250mm×4.6mm,5μm)、Eclipse XDB-phenyl(250mm×4.6mm,5μm)以及Xbridge-C18柱(250mm×4.6mm,5μm)、AQ-C18(250 mm×4.6mm,5μm)、Diamonsil -C18(150mm×4.6mm,5μm)、Weltch XB-C18(150mm×4.6mm,5μm)、Proshell EC C18(150mm×4.6mm,5μm)等不同色谱柱对p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚、溴氯芬及六氯酚等六种组分和样品基质的分离效果。结果显示,上述色谱柱对标准溶液中目标化合物都可以达到良好的分离效果,但在实际样品分析时,Weltch XB-C18(150mm×4.6mm,5μm)和Eclipse XDB-phenyl(150mm×4.6mm,5μm)对大部分样品的分离效果较好,由于溴氯芬及六氯酚在色谱柱上难以洗脱,应避免使用含碳量高的色谱柱。鉴于C18色谱柱通用性好,本方法选定Weltch XB-C18(150mm×4.6mm,5μm)为分析色谱柱。
2.3 流动相的选择
六种氯酚分子结构中的羟基和色谱柱固定相中残留的硅羟基存在较强的氢键作用,使化合物在色谱柱上保留较强,因此,若仅以甲醇-水或乙腈-水作为流动相,可能会出现色谱峰严重拖尾或不出峰的情况。然而,加入甲酸等改性剂,可以有效减小拖尾现象。因此,本方法考察了甲醇-0.1%甲酸水和乙腈-0.1%甲酸水两种流动相体系。实验结果表明,在0.1%甲酸-乙腈流动相体系中,六种氯酚的分离度好,峰形对称,灵敏度高。因此,本方法选用0.1%甲酸-乙腈作为流动相。此外,六种目标物极性差异较大,不适合采用等度洗脱,而使用梯度洗脱时,随时间增加逐渐增加有机相比例,不仅可以加快目标物的洗脱,还可以减小半峰宽。优化后的六种氯酚标准物质色谱图见图1和图2。

图1 标准物质溶液的液相色谱图(λ=282nm)
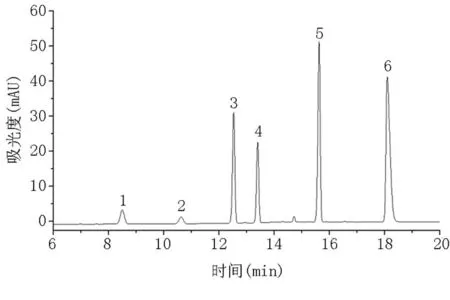
图2 标准物质溶液的液相色谱图(λ=298nm)
2.4 检测波长的选择
在200~400nm 范围内对六种氯酚的标准溶液进行全波长扫描,分别获得相应的紫外吸收光谱图(图3所示)。


图3 六种标准物质溶液的紫外吸收光谱图
由表1可知,p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚、溴氯芬和六氯酚的质量浓度在0.5~20μg/mL范围内与相应色谱峰面积呈良好的线性关系,六种氯酚的方法检出限为4μg/g,方法定量限为10μg/g。

表1 六种目标物的线性方程、线性范围、相关系数、方法检出限和定量限
2.5 线性方程和检出限
在1.4仪器条件下对系列标准工作溶液进行测定。以各目标物质量浓度(x)为横坐标,对应峰面积(y)为纵坐标,分别绘制标准工作曲线。对阴性样品添加适量六种氯酚混合标准溶液,按照试样前处理方法和仪器条件进行测定,以信噪比S/N≥3确定方法检出限,以信噪比S/N≥10确定方法定量限。线性范围、线性方程、相关系数、检出限和定量限见表2。
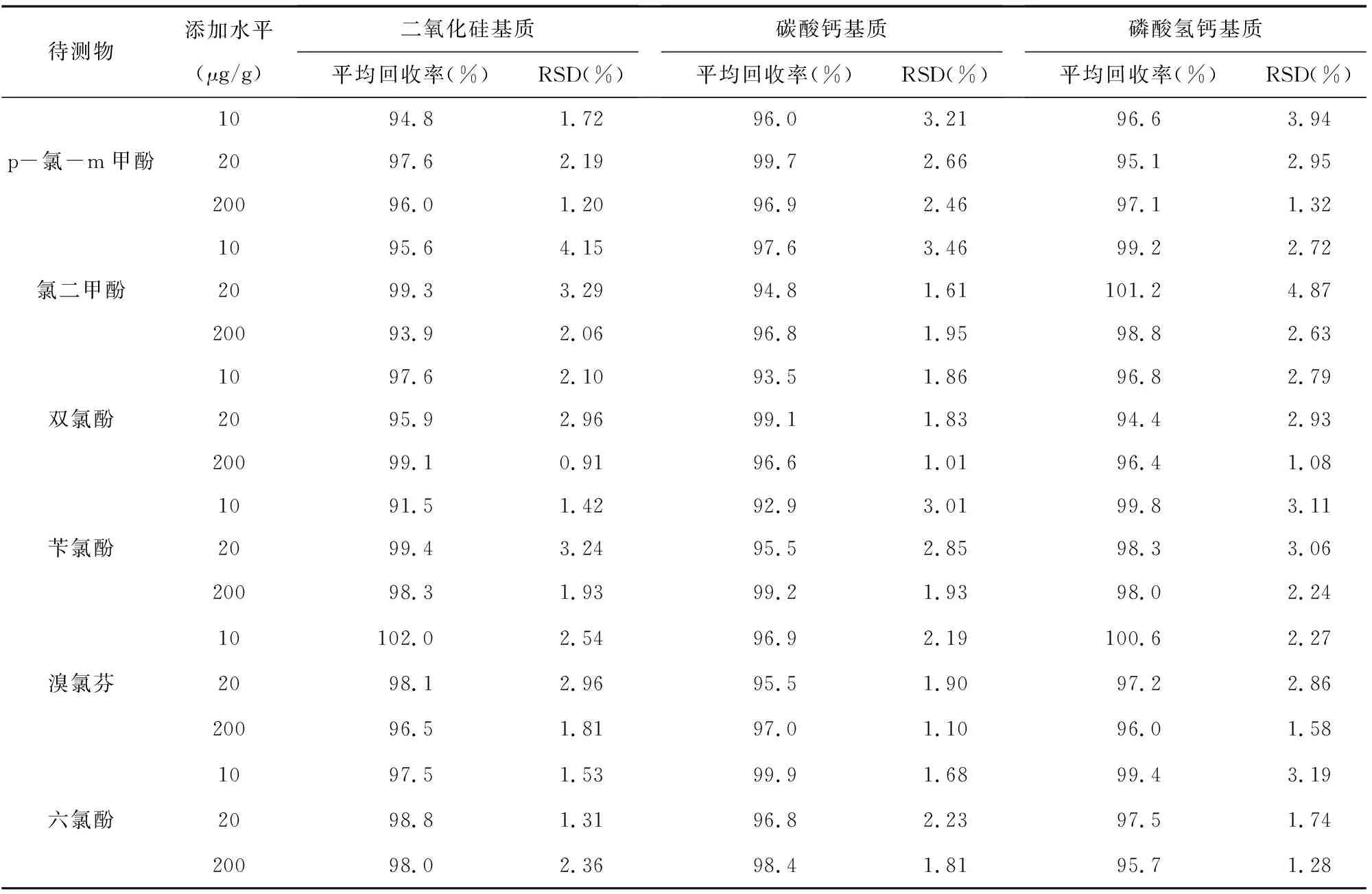
表2 精密度试验和加标回收试验结果(n=6)
2.6 精密度试验及加标回收试验
选用二氧化硅基质、碳酸钙基质和磷酸氢钙基质3种空白牙膏样品为加标基质,按1.3方法进行处理,分别加入低、中、高3个加标水平的氯酚标准溶液(添加水平分别为10μg/g、20mg/kg、200μg/g),在1.4仪器条件下平行测定6次,测定结果见表2。由表2可知,p-氯-m甲酚的平均回收率为94.8%~99.7%,相对标准偏差为1.20%~3.94%(n=6);氯二甲酚的平均回收率为93.9%~101.2%,相对标准偏差为1.61%~4.87%(n=6);双氯酚的平均回收率为93.5%~99.1%,相对标准偏差为0.91%~2.96%(n=6);苄氯酚的平均回收率为91.5%~99.8%,相对标准偏差为1.42%~3.24%(n=6);溴氯芬的平均回收率为95.5%~102.0%,相对标准偏差为1.10%~2.96%(n=6);六氯酚的平均回收率为95.7%~99.9%,相对标准偏差为1.28%~3.19%(n=6)。结果表明,本方法具有良好的准确度和精密度,满足分析要求。
3 结论
本研究采用高效液相色谱技术建立了牙膏中p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚、溴氯芬及六氯酚的检测方法。对提取条件和色谱条件等进行了优化,并将该方法应用于不同基质牙膏的检测。结果表明,该方法能对p-氯-m-甲酚、氯二甲酚、双氯酚、苄氯酚、溴氯芬及六氯酚进行准确测定,且方法精密度好,满足相关检测需求,可为牙膏产品的质量监控提供有力的技术支撑。
猜你喜欢
中老年保健(2022年4期)2022-11-25 14:45:02
煤化工(2022年3期)2022-07-08 07:24:42
环境污染与防治(2020年7期)2020-07-27 07:31:38
小猕猴学习画刊(2016年12期)2017-01-05 21:38:26
作文评点报·低幼版(2016年16期)2016-05-30 09:57:42
学苑创造·A版(2016年1期)2016-03-10 18:09:10
中国资源综合利用(2016年10期)2016-01-22 08:36:09
中国药业(2014年17期)2014-05-26 09:07:45
长江大学学报(自科版)(2013年33期)2013-03-11 15:08:22
化工技术与开发(2011年6期)2011-12-18 06:29:56
