
Potential Secretory Transporters and Biosynthetic Precursors of Biological Nitrification Inhibitor 1,9-Decanediol in Rice as Revealed by Transcriptome and Metabolome Analyses
2024-03-09 08:45DiDongweiMaMingkunZhangXiaoyangLuYufangHerbertKronzuckerShiWeiming
Rice Science 2024年1期
Di Dongwei, Ma Mingkun, , Zhang Xiaoyang, Lu Yufang, Herbert J. Kronzucker, Shi Weiming, 2,
Research Paper
Potential Secretory Transporters and Biosynthetic Precursors of Biological Nitrification Inhibitor 1,9-Decanediol in Rice as Revealed by Transcriptome and Metabolome Analyses
Di Dongwei1, Ma Mingkun1, 4, Zhang Xiaoyang1, Lu Yufang1, Herbert J. Kronzucker3, Shi Weiming1, 2,4
(; School of BioSciences, The University of Melbourne, Parkville, VIC 3010, Australia; )
Biological nitrification inhibitors (BNIs) are released from plant roots and inhibit the nitrification activity of microorganisms in soils, reducing NO3‒leaching and N2O emissions, and increasing nitrogen- use efficiency (NUE). Several recent studies have focused on the identification of new BNIs, yet little is known about the genetic loci that govern their biosynthesis and secretion. We applied a combined transcriptomic and metabolomic analysis to investigate possible biosynthetic pathways and transporters involved in the biosynthesis and release of BNI 1,9-decanediol (1,9-D), which was previously identified in rice root exudates. Our results linked four fatty acids, icosapentaenoic acid, linoleate, norlinolenic acid, and polyhydroxy-α,ω-divarboxylic acid, with 1,9-D biosynthesis and three transporter families, namely the ATP-binding cassette protein family, the multidrug and toxic compound extrusion family, and the major facilitator superfamily, with 1,9-D release from roots into the soil medium. Our finding provided candidates for further work on the genes implicated in the biosynthesis and secretion of 1,9-D and pinpoint genetic loci for crop breeding to improve NUE by enhancing 1,9-D secretion, with the potential to reduce NO3‒leaching and N2O emissions from agricultural soils.
1,9-decanediol; biological nitrification inhibitor; metabolomic analysis; nitrogen-use efficiency; transcriptomic analysis
The two main sources of nitrogen (N) utilized by higher plants are ammonium-N (NH4+) and nitrate-N (NO3‒). Nitrification, a process carried out by soil microorganisms, can readily convert NH4+to NO3‒,but the extent of this conversion varies greatly among soil environments (Kronzucker et al, 1997, 1999; Kaur- Bhambra et al, 2022). Unlike the cationic NH4+form, which is retained well in most soil types, the anionic NO3‒form can leach from soils into water systems and cause environmental pollution. Denitrification produces various gaseous forms of N, including N2O, NO, and N2, with N2O being a potent greenhouse gas and resulting in global warming (Coskun et al, 2017b; Min and Shi, 2018; Min et al, 2021; Souza et al, 2021; Li et al, 2022; Elrys et al, 2023). Nitrification inhibitors, including both synthetic and biological nitrification inhibitors (BNIs), inhibit nitrification by soil bacteria and thereby reduce NO3‒loss and N2O emission while improving nitrogen-use efficiency (NUE) (Subbarao et al, 2013b, c; Coskun et al, 2017a, b). In contrast to the typically lower efficacy and higher environmental pollution potential of synthetic nitrification inhibitors, BNIs released from plant roots possess high efficacy and are more environmentally friendly (Subbarao et al, 2015; Woodward et al, 2016).
Over the past two decades, several BNIs, including methyl 3-(4-hydroxyphenyl) propionate, sakuranetin, sorgoleone, brachialactone, syringic acid, 2,7-dimethoxy-1,4-naphthoquinone, 2-hydroxy-4,7-dimethoxy-2H-1,4- benzoxazin-3(4H)-one, 6-methoxy-2(3H)-benzoxazolone,oxalic acid, and protocatechuic aldehyde, have been identified from,,,,,, and(Zakir et al, 2008; Subbarao et al, 2009, 2013a; Sun et al, 2016; Lu et al, 2022; Otaka et al, 2022;Wang et al, 2023). A practical approach to promoting NUE involves crop rotation, where crops that can secrete BNIs from their roots are alternated with those that cannot or by mixing crude extracts from BNI-secreting plants with standard fertilizers (Wang et al, 2023). For example, rotations of sorghum and vegetables can effectively reduce N2O emissions and increase NUE in vegetable crops, whileextracts have been shown to simultaneously inhibit soil urease activity, soil nitrification, and urea degradation, resulting in greatly improved NUE in potato tubers (Zhang et al, 2015; Elrys et al, 2019). However, the biosynthetic pathways of BNIs and their regulation are, in most cases, poorly understood. Recently, Wang et al (2023) employed a multi-omic analysis to infer the biosynthetic pathways of protocatechuic aldehyde and oxalic acid inMore generally, identifying the pathways of BNI synthesis and targeting these by genetic engineering to improve BNI secretion or adding BNI-secretion capabilities to plants that cannot secrete BNIs would be a potent means to decrease NO3‒leaching and N2O emissions from crop systems and improve the NUE of crops (Subbarao et al, 2021).
Recently, we identified a new BNI from rice root exudates, 1,9-decanediol (1,9-D), a fatty alcohol compound that inhibits nitrification by blocking ammonia monooxygenase (AMO) in soil bacteria (Sun et al, 2016). A subsequent study showed that the release of 1,9-D from rice roots is stimulated by NH4+, pH modification, and nitrifying bacteria (Zhang et al, 2019). Further research demonstrated that 1,9-D inhibits nitrification through its action on both ammonia- oxidizingbacteria and archaea in soils (Lu et al, 2019). In addition, our more recent study inrevealed that 1,9-D promotes primary root growth by regulating abscisic acid content and PIN2-dependent auxin transport (Ma et al, 2023). Although past studies have increased our understanding of 1,9-D, the mechanisms of its synthesis and release from roots remained unclear.
In recent years, multi-omic analysis has been increasingly used as an effective approach to identifying gene networks and regulatory pathways in a large variety of organisms (Fu et al, 2021; Wang et al, 2021). An integrated analysis of both the transcriptome and the metabolome has been deployed to uncover metabolic pathways in response to specific environmental and developmental signals (Cheng et al, 2022; Zhang Y T et al, 2022; Wang et al, 2023). In the present study, we utilized a combined transcriptome and metabolome analysis of two rice varieties, Wuyunjing 7 (WYJ7, high 1,9-D release) and Wuyunjing 3 (WYJ3, no 1,9-D release), as well as NH4+-treated WYJ7, to identify the possible metabolic pathways involved in 1,9-D biosynthesis. We also screened for possible genes encoding 1,9-D secretion carriers and/or channels by analyzing up-regulated genes related to transport carriers and/or channels in RNA-seq data. Our study sheds light on the genetic loci of 1,9-D synthesis and secretion and provides possible gene targets for screening rice varieties with high BNI secretion capacity. This has the promise of reducing N losses from rice systems and improving NUE in this major crop species.
Results
Rice varieties and rhizospheric NH4+ influence 1,9-D release
To identify key genes affecting the release of 1,9-D in rice, we performed a combined analysis of the transcriptome and metabolome. Our previous studies showed that N form (NH4+or NO3‒), concentration, pH, aeration, bacterial inoculum (nitrifying or denitrifying bacteria), and rice variety all have an effect on 1,9-D release. Of these, NH4+and varietal differences have a particularly pronounced effect on 1,9-D release (Sun et al, 2016; Zhang et al, 2019). In the current study, we first tested for 1,9-D release prior to transcriptomic and metabolomic analyses. Consistent with previous results, our data showed that external NH4+, supplied as NH4Cl at 1 mmol/L, increased 1,9-D release in WYJ7 by 74% (Fig. 1-A). In contrast, we did not detect any 1,9-D release in WYJ3 grown under control conditions, even after NH4+treatment (Fig. 1-B). These clear differences in 1,9-D release between rice varieties and in response to NH4+treatment provided an excellent framework within which to explore transcriptomic and metabolomic analyses.

Fig. 1. Influence of NH4+and variety on 1,9-decanediol (1,9-D) release in rice.
A, 1,9-D release from Wuyunjing 7 (WYJ7) roots with or without 1 mmol/L NH4Cl (NH4+) treatment.
B, 1,9-D release in Wuyunjing 3 (WYJ3) and WYJ7 grown in control media.
Six-week-old seedlings were transferred to new solutions with 1 mmol/L CaCl2(control) or 1 mmol/L NH4Cl (NH4+treatment) and grown for another 24 h prior to exudate collection. ND indicates not detected. Data are Mean ± SD with four replications. ***,< 0.001 (test).
Gas chromatography-mass spectrometer (GC-MS)- based metabolomics reveals distinct changes in metabolites in two experimental groups
We first used GC-MS to identify differentially synthesized metabolites (DSMs) in two experimental groups, NH4+-WYJ7 vs CK-WYJ7 (N7/C7) and CK-WYJ7 vs CK-WYJ3 (C7/C3).Orthogonal partial least squares-discriminant analysis (OPLS-DA) divided all samples into different clusters, demonstrating that rice variety and NH4+treatment produced clear differences in metabolite accumulation (Fig. 2-A and -B). To prevent overfitting of the model, we further performed response permutation testing, and R2 values were 0.945 and 0.974 in N7/C7 and C7/C3, respectively, underscoring the reliability of the OPLS- DA models (Fig. S1). A one-dimensional analysis was used to screen DSMs in the two groups, and 38 and 14 DSMs were identified in the N7/C7 and C7/C3 groups, respectively (Fig. 2-C and -D; Table S1). Among these DSMs, l-sorbose, 3-phosphoglyceric acid, d-mannose 6-phosphate, and galactinol were enriched more than 20-fold in WYJ7 after the NH4+treatment. However, compared with C3, only two DSMs, vanillic acid and terephthalic acid, were up- regulated in C7 (Table S1). Subsequently, we further classified the DSMs by the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. Compared with C3, the DSMs in C7 were significantly (< 0.05) enriched in the ATP-binding cassette (ABC) transporters, glutathione metabolism, and arginine biosynthesis. In comparison, the DSMs were significantly (< 0.05) enriched in alanine, aspartate and glutamate metabolism, aminoacyl-tRNA biosynthesis, and glyoxylate and dicarboxylate metabolism in N7 compared with those in C7 (Fig. 2-E and -F; Table S2).
Liquid chromatography-mass spectrometer (LC-MS)-based metabolomics reveals distinct changes in metabolites in two experimental groups
To further examine the DSMs in different rice varietiesand under NH4+treatment, we analyzed the non-targeted metabolome of roots using LC-MS. We identified 735 and 508 DSMs (< 0.05) in the N7/C7 and C7/C3 groups, respectively (Table S3). OPLS-DA model analyses revealed distinct clusters between C7 and N7, and/or C3, indicating the presence of significantly different metabolites in the N7/C7 and C7/C3 groups (Fig. 3-A and -B). In the N7/C7 group, we obtained 481 up-regulated and 254 down-regulated DSMs, which were enriched in 164 pathways, including 79 significant metabolic pathways (< 0.05) (Fig. 3-C and -D; Table S4). The up-regulated DSMs were significantly enriched in linoleic acid metabolism, arachidonic acid metabolism, and glycerophospholipid metabolism, while the down-regulated DSMs were significantly enriched in the citrate cycle (TCA cycle), histidine metabolism, and glyoxylate and dicarboxylate metabolism (Figs. 3-D and S2). Moreover, in the C7/C3 group, we identified 43 up-regulated and 465 down- regulated DSMs, which were further classified into 52 metabolic pathways, of which 27 pathways were significant (< 0.05) (Fig. 3-E and -F; Table S4). The up-regulated DSMs in the C7/C3 group were significantly enriched in linoleic acid metabolism, arachidonic acid metabolism, and pantothenate and CoA biosynthesis, while the down-regulated DSMs were significantly enriched in ABC transporters, glutathione metabolism, and galactose metabolism (Figs. 3-F and S3). Although there were differences in the metabolic pathways enriched in the N7/C7 and C7/C3 groups, the common up- or down-regulation of metabolic pathways also implies that the metabolic pathway responsible for 1,9-D biosynthesis in WYJ7 may be implicated in both the response to NH4+and underpin the differences between rice varieties.
RNA-seq analysis reveals distinct changes in transcriptome in two experimental groups
To identify potential genes involved in regulating 1,9-D biosynthesis and secretion, we sequenced nine samples(C7, N7, and C3, with three replicates each) using RNA-seq technology. The numbers of raw and clean reads are shown in Table S5. The total mapped reads were more than 90% (Table S5). We found that 800 and 1 308 differentially expressed unigenes (DEGs) were up-regulated, while 832 and 2 153 DEGs were down- regulated in N7/C7 and C7/C3 groups, respectively (Table S6). Additionally, 90 (24 at< 0.05) and 91 (17 at<0.05) Gene Ontology (GO) terms tested for N7/C7 and C7/C3 were divided into three categories, biological process, cellular component, and molecular function. The largest subcategory under biological process was metabolic process, while cell and catalytic activity were the largest subcategories under cellular component and molecular function, respectively (Fig. 4-A and -B; Table S7). Using KEGG enrichment analysis, weidentified the biosynthetic pathways of bioactive components in N7/C7 and C7/C3. We found 477 and 730 DEGs were annotated into five categories, namely cellular process, environmental information processing,genetic information processing, metabolism, and organismal systems, which were further divided into 18 and 18 subcategories, respectively (Fig. 4-C and -D; Table S8). Of these subcategories, carbohydrate metabolism (60/135), lipid metabolism (56/64), amino acid metabolism (49/85), biosynthesis of other secondary metabolites (56/68), and signal transduction (43/49) were correlated to more DEGs compared with the other subcategories in N7/C7 and C7/C3, respectively (Table S8).
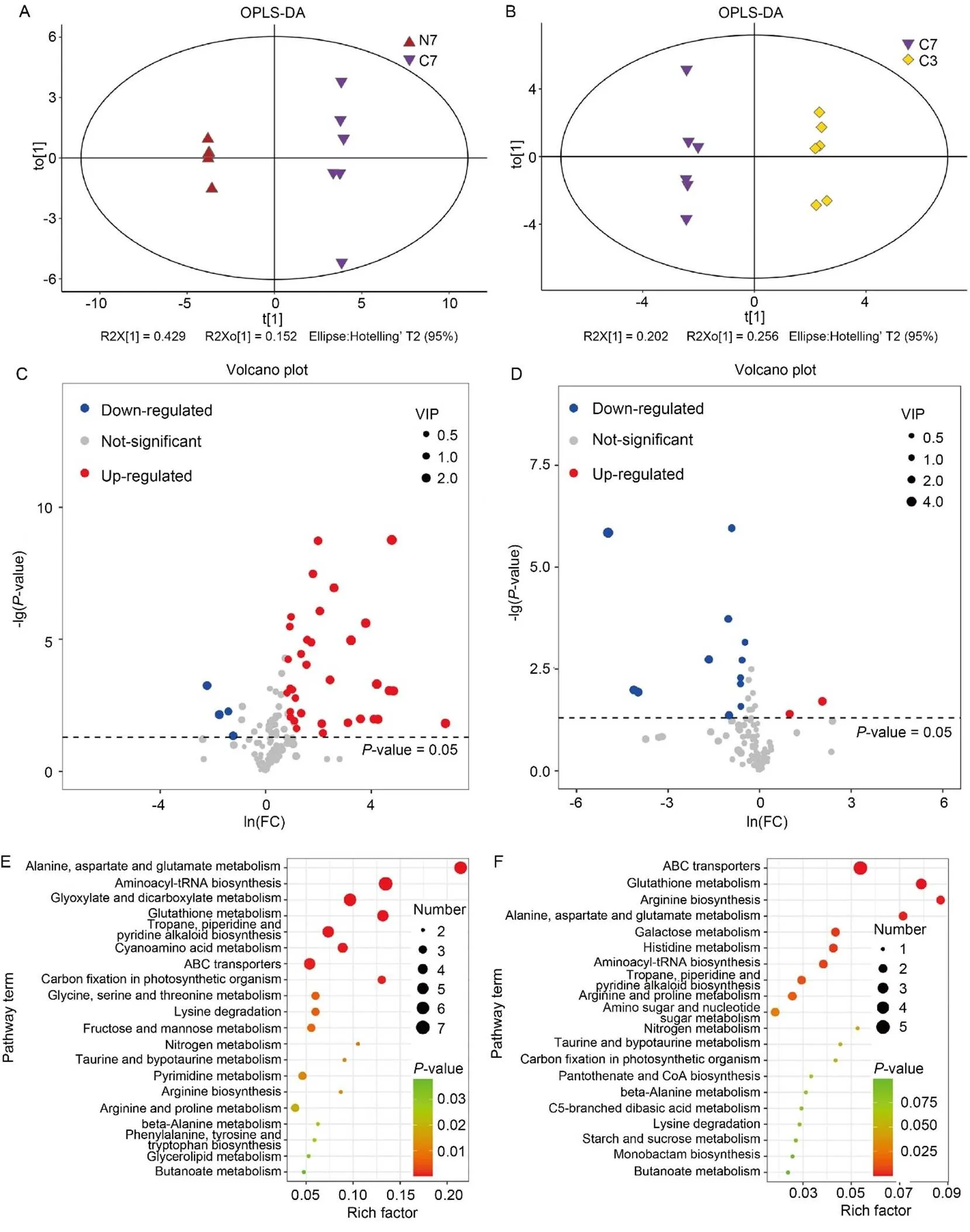
Fig. 2. Analysis of differentially synthesized metabolites (DSMs) based on gas chromatography-mass spectrometer (GC-MS).
A and B, Orthogonal partial least squares-discriminant analysis (OPLS-DA) of NH4+-Wuyunjing 7 (N7)/CK-Wuyunjing 7 (C7) (A) and C7/CK- Wuyunjing 3 (C3) (B) groups.
C and D, Volcano plots of DSMs in N7/C7 (C) and C7/C3 (D) groups. Each point in the volcano plot indicates an identified metabolite, and the red/blue/grey dots indicate up-/down-/unregulated metabolites. VIP, Variable importance of projection.
E and F, Kyoto Encyclopedia of Genes and Genomes analysis of DSMs in N7/C7 (E) and C7/C3 (F) groups. The size and color of the dots indicate the number of DSMs and the significance (< 0.05), respectively. ABC, ATP-binding cassette.

Fig. 3. Analysis of differentially synthesized metabolites (DSMs) based on liquid chromatography-mass spectrometer (LC-MS).
A and B, Orthogonal partial least squares-discriminant analysis (OPLS-DA) of NH4+-Wuyunjing 7 (N7)/CK-Wuyunjing 7 (C7) (A) and C7/CK- Wuyunjing 3 (C3) (B) groups.
C and D, Volcano plots of DSMs in N7/C7 (C) and C7/C3 (D) groups. Each point in the volcano plot indicates an identified metabolite, and the red/blue/grey dots indicate up-/down-/unregulated metabolites. VIP, Variable importance of projection.
E and F, Kyoto Encyclopedia of Genes and Genomes analysis of DSMs in N7/C7 (E) and C7/C3 (F) groups. The size and color of the dots indicate the number of DSMs and the significance (< 0.05), respectively. ABC, ATP-binding cassette.

Fig. 4. Analysis of differentially expressed unigenes (DEGs) based on RNA-seq analysis.
A and B, Gene Ontology (GO) classification of DEGs in NH4+-Wuyunjing 7 (N7)/CK-Wuyunjing 7 (C7) (A) and C7/CK-Wuyunjing 3 (C3) (B) groups.
C and D, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DEGs in N7/C7 (C) and of C7/C3 (D) groups.
E, Venn diagram showing co-regulated DEGs in N7/C7 and C7/C3 groups.
F, Hierarchical clustering heatmap of co-upregulated DEGs in N7/C7 and C7/C3 groups.
To further identify common DEGs in the N7/C7 and C7/C3 groups, we performed Venn analysis and calculated the co-upregulated and co-downregulated DEGs (Fig. 4-E). There were 55 co-downregulated and 47 co-upregulated DEGs in the N7/C7 and C7/C3 groups. In our effort to identify genes positively associated with enhanced 1,9-D release, we focused mainly on the co-regulated genes. The GO test suggested that these 47 co-upregulated DEGs were mainly associated with catalytic activity, metabolic process, and response to stress (Fig. 4-E; Table S9). Furthermore, the KEGG enrichment analysis showed that these DEGs were distributed across several pathways, including diterpenoid biosynthesis, fatty aciddegradation-linolenic acid metabolism, phenylpropanoid biosynthesis, and fructose and mannose metabolism (Fig. 4-F; Table S9).
Integrated analysis of metabolome and transcriptome in two experimental groups
To further examine potential target genes, we performed a Spearman’s correlation analysis between DEGs and DSMs in the N7/C7 and C7/C3 groups (Fig. 5-A and -B). To clarify which DEGs were co-regulated in N7/C7 and C7/C3, we conducted Venn analysis and identified 1 co-upregulated gene and 1 co-down- regulated gene among the 13 co-regulated DEGs (Fig. 5-C). The co-upregulated DEGencodes an uncharacterized protein belonging to the inhibitor I protein family, while the co-downregulated DEGencodes an unknown protein (Table S10). To further explore which DSMs were responsible for the response to NH4+treatment and rice variety differences, we first screened for up-regulated DSMs that were identified as associated DEGs in the N7/C7 and C7/C3 groups via Venn analysis. We identified 22 and 20 up-regulated DSMs with correlated DEGs in the N7/C7 and C7/C3 groups, respectively (Fig. 5-D). In a further Venn analysis of these DSMs, we screened for five co-upregulated DSMs, namely 9-oxo-13-hydroxy-11-octadecenoic acid, (9R,12R,15R)-d10-13-PhytoF[13R,16R], (10R,13R,16R)-d14-9-PhytoF[9S,12S], benzarone, and trimethobenzamide (Fig. 5-D). Of these five DSMs, 9-oxo-13-hydroxy- 11-octadecenoic acid, (9R,12R,15R)-d10-13-PhytoF [13R,16R], and (10R,13R,16R)-d14-9-PhytoF[9S,12S] were all categorized within octadecanoids (Table S2).

Fig. 5. Identification of co-regulated differentially expressed unigenes (DEGs) and differentially synthesized metabolites (DSMs) via integrated analysis of metabolome and transcriptome.
A and B, Correlation analysis of top 20 DEGs and DSMs in NH4+-Wuyunjing 7 (N7)/CK-Wuyunjing 7 (C7) (A) and C7/CK-Wuyunjing 3 (C3) (B) groups. Red/blue represents a positive/negative correlation. *, **, and *** represent 0.05, 0.01, and 0.001 levels, respectively.
C, Venn analysis of co-regulated DEGs of top 100 correlation between DEGs and DSMs in N7/C7 and C7/C3 groups.
D, Venn analysis of co-upregulated DSMs in N7/C7 and C7/C3 groups.
Identification of possible metabolites and their associated genes in rice
We then analyzed the boxcharts of three selected DSMs and found that the relative levels of these three DSMs were the least in CK-WYJ3 and the highest in NH4+-WYJ7, consistent with the level of 1,9-D, revealing the potential role of octadecanoids during 1,9-D biosynthesis (Figs. 1 and 6-A). To identify the related genes involved in the biosynthesis of these three DSMs, we screened the associated genes from ‘DSMs correlation to DEGs’ data and found four up- regulated genes,,,, and, in the N7/C7 and C7/C3 groups (Table S10).
To verify the reliability of the RNA-seq data, we further analyzed the transcriptional levels of genes responsible for these three DSMs by qRT-PCR. The transcription patterns of the selected genes were similar to the RNA-seq analysis, indicating that the data obtained from RNA-seq were reliable (Fig. 6-B).
Screening of possible genes encoding 1,9-D secretion vectors by RNA-seq data
In addition to biosynthesis ability, the amount of secretory carrier also affects 1,9-D release. To identify potential genes involved in regulating 1,9-D release, we first screened for up-regulated transporter-related DEGs in the N7/C7 and C7/C3 groups. We found 40 and 22 DEGs in the N7/C7 and C7/C3 groups, respectively (Fig. 7-A and -B). Functional description revealed that these DEGs were divided into several transporter families, such as the ABC protein family, the multidrug and toxic compound extrusion (MATE) family, and the peptide transporter family (Table S11). Further Venn analysis identified three co-upregulated genes in both the N7/C7 and C7/C3 groups:,, and(Fig. 7-C). Subsequent qRT-PCR analysis confirmed that the transcription levels of these genes were induced by NH4+treatment in WYJ7 and inhibited in C3 compared with C7 (Fig. 7-D).

Fig. 6. Identification of possible genes involved in co-upregulated differentially expressed unigenes (DEGs) via qRT-PCR.
A, Boxchart of three co-upregulated metabolites.
B, qRT-PCR analysis of possible genes associated to DEGs. Six-week-old rice seedlings are transferred to fresh solutions with 1 mmol/L CaCl2(control) or 1 mmol/L NH4Cl (NH4+treatment) and grown for another 24 h prior to RNA extraction of roots. All data are normalized relative to().
N7, C7, and C3 represent NH4+-Wuyunjing 7, CK-Wuyunjing 7, and CK-Wuyunjing 3, respectively. Values indicate Mean ± SD of three biological replicates. Error bars with different lowercase letters represent statistically significant differences (< 0.05, Duncan’s test).

Fig. 7. Identification of possible 1,9-decanediol secretion-related genes.
A and B, Hierarchical clustering heatmap of co-upregulated transporter-related differentially expressed unigenes (DEGs) in NH4+-Wuyunjing 7 (N7)/CK-Wuyunjing 7 (C7) (A) and C7/CK-Wuyunjing 3 (C3) (B) groups.
C, Venn analysis of co-upregulated transporter-related genes in N7/C7 and C7/C3 groups.
D, qRT-PCR analysis of co-upregulated genes. Six-week-old rice seedlings are transferred to fresh solutions with 1 mmol/L CaCl2(control) or 1 mmol/L NH4Cl (NH4+treatment) and grown for another 24 h prior to RNA extraction of roots. All data are normalized relative to(). Values indicate Mean ± SD of three biological replicates. Error bars with different lowercase letters represent statistically significant differences (< 0.05, Duncan’s test).
Discussion
BNIs released by roots can effectively suppress bacterial nitrification in a variety of soils, including agricultural soils. This leads to the stabilization of the NH4+form of N, allowing plants to use NH4+directly and reducing N loss from soils through NO3‒leaching and N2O emissions (Subbarao et al, 2006; Coskun et al, 2017b; Zhang M X et al, 2022). The identification and functional analysis of BNIs have been successful in sorghum, pasture grasses, rice, maize, and wheat(Subbarao et al, 2007, 2009; Zakir et al, 2008; O’Sullivan et al, 2016; Sun et al, 2016). BNIs released from the roots of these plants decrease the abundance of both ammonia-oxidizing bacteria and archaea in the rhizosphere, greatly reducing overall soil-bacterial nitrification(Lu et al, 2019; Nardi et al, 2020). However, it remains unclear how BNIs are synthesizedand by what mechanisms they are secreted into the rhizosphere(Zhang M X et al, 2022). Given the pronounced differences in 1,9-D secretion in different rice varieties (WYJ3 and WYJ7) and in response to NH4+treatment (Fig. 1), we have used this as a basis for a multi-omics screen, pinpoint possible biosynthetic pathways and transporters that may be involved in 1,9-D secretion.
Are ‘fatty acids’ pathways responsible for 1,9-D biosynthesis in rice?
Our previous study showed that 1,9-D is a C10 fatty alcohol (Sun et al, 2016). Fatty alcohols consist of a non-polar, lipophilic carbon chain (C8‒C18) and a polar, hydrophilic hydroxyl group. In engineered microbes, the biosynthesis of fatty alcohols begins with the formation of fatty acyl-acyl carrier proteins (FA-ACPs) (Zheng et al, 2012). Subsequently, FA-ACPs are converted to free fatty acids and fatty acyl coenzyme A (FA-CoA) through a series of reactions catalyzed by thioesterase and acyl-CoA synthase (Lu et al, 2008). Thereafter, FA-CoA is reduced to fatty aldehydes or fatty alcohols in an NADPH-dependent manner, catalyzed by fatty acyl-CoA reductase. In turn, fatty aldehydes can also be converted to fatty alcohols by alcohol dehydrogenases or aldehyde reductases (Reiser and Somerville, 1997; Vioque and Kolattukudy, 1997). Hence, thioesterase, acyl-CoA synthase, and fatty acyl-CoA reductase are critical enzymes in fatty alcohol biosynthesis(Steen et al, 2010). Interestingly, these enzymes differ significantly in substrate specificity (Liu et al, 2011).
To identify metabolites and genes positively associated with 1,9-D biosynthesis, we performed a Spearman correlation analysis between DEGs and DSMs in the N7/C7 and C7/C3 groups, selecting the top 20 for further investigation (Fig. 5-A and -B). KEGG enrichment analysis revealed that these DEGs and DEMs were enriched in 58 and 49 metabolic pathways in the N7/C7 and C7/C3 groups, respectively, with 42 metabolic pathways being co-enriched in both groups (Table S10). A Venn analysis identified the co- upregulated DEGs and revealed 13 co-regulated DEGs, of which only one DEG was co-upregulated and one DEG was co-downregulated in both the N7/C7 and C7/C3 groups (Fig. 5-C). Subsequent ‘Gene Function Annotation’ suggested that these two DEGs may not be potential genes related to 1,9-D biosynthesis (Table S9). Further screening of the co-upregulated DSMs in the N7/C7 and C7/C3 groups identified five co- upregulated DSMs (Fig. 5-D). Of these five DSMs, 9-oxo-13-hydroxy-11-octadecenoic acid, (9R, 12R,15R)-d10-13-PhytoF[13R,16R], and (10R,13R,16R)-d14-9- PhytoF[9S,12S] fall into the category of linoleic acid metabolism (osa00591), and more broadly, with the metabolism of octadecanoids, i.e. C18 fatty acid (Fig. 5-D; Table S8). Octadecanoids have been described as signaling compounds induced under stress, such as in response to wounding and as part of plant defense(Fliegmann et al, 2003). However, it is unclear whether the octadecanoids involved in such responses are also the synthetic precursors of 1,9-D. Our data indicated that three metabolites associated with octadecanoid metabolism and their associated genes were distinctly up-regulated in both N7/C7 and C7/C3 (Fig. 6).
We further analyzed the enriched metabolic pathways (KEGG enrichment) through a top 100 correlation analysis (< 0.05). The DSMs involved in cutin, suberin, and wax biosynthesis (osa00073), α-linolenic acid metabolism (osa00592), and the biosynthesis of unsaturated fatty acids (osa01040), all of which are implicated in fatty acid metabolism, were up-regulated in the C7/C3 group, but not in the N7/C7 group. This indicated that thedifferences in 1,9-D content between the two rice varieties (WYJ7 vs WYJ3) and the differences in 1,9-D content following NH4+application (NH4+vs CK in WYJ7) were accomplished via different metabolic pathways (Fig. 8; Table S8). In the context of the aforementioned DSMs, it is instructive to point out that linolenic acid and linoleate are also isolated from and identified inshoots as BNIs. These compounds are capable of inhibiting the activities of AMO and hydroxylamino oxidoreductase in(Subbarao et al, 2008). Additionally, the fatty acids, palmitic acid and oleic acid, which may also participate in 1,9-D biosynthesis, have been reported to block AMO activity inat higher concentrations(Subbarao et al, 2008) (Fig. 8). These findings suggest important roles of these plant- metabolic pathways in inhibiting bacterial nitrification.
Much work remains to definitively establish which metabolic pathway components are involved in the biosynthesis of 1,9-D, at both the biochemical and genetic levels. To achieve this, the following steps may be effective: (i) exogenous application of candidate metabolites to rice roots, coupled with the determination of the amount of 1,9-D exudation in response to each application; (ii) application of CRISPR/Cas9 and/or gene overexpression techniques to knockout or up-regulate genes in relevant metabolic pathways, coupled with the determination of the amount of 1,9-D exudation in the transgenic materials.

Fig. 8. Potential biosynthesis pathways and transporters responsible for 1,9-decanediol (1,9-D) synthesis and release in rice.
A, Potential metabolic pathways involved in 1,9-D biosynthesis.
B, Potential transporters responsible for 1,9-D release.
ABC, ATP-binding cassette protein family; MATE, Multidrug and toxic compound extrusion family; MFS, Major facilitator superfamily. DSMs, Differentially synthesized metabolites; DEGs, Differentially expressed unigenes.
Which transporters are responsible for transport of 1,9-D in rice roots?
BNIs can be divided into hydrophilic and hydrophobic BNIs, according to the compounds’ solubility in water(Zhang M X et al, 2022). For hydrophobic BNIs, such as sorgoleone, the release from roots can occur through exocytosis and/or vesicle trafficking (Czarnota et al, 2003). By contrast, the release of hydrophilic BNIs may be mediated by voltage-gated anion channels and/or transporters(Zhang M X et al, 2022). To identify the carriers or channels responsible for the release of 1,9-D into the rhizosphere, we performed RNA-seq analysis on CK-WYJ3 (no 1,9-D release), CK-WYJ7 (high 1,9-D release), and NH4+-WYJ7 (higher 1,9-D release) roots. We then identified up-regulated DEGs categorized under the ‘transporter ability’ GO item, in the two groups, C7/C3 and N7/C7 (Fig. 7; Table S11).
We screened 58 and 40 up-regulated DEGs in the C7/C3 and N7/C7 groups, respectively (Fig. 7-A and -B). Venn analysis then revealed that the two groups shared three DEGs in common, namely,, and(Fig. 7-C).encodes a CorA-like magnesium transporter protein, which may possess the same magnesium transport capacity as its homologous protein LOC_Os01g64890 (OsMGT1) (Chen et al, 2012). LOC_Os03g22200 (OsSWEET16), a nodulin MtN3 family protein, functions as a sugar transporter and is involved in responses to plant pathogen infection (Streubel et al, 2013). The protein encoded bybelongs to a family of transmembrane amino acid transport proteins, of which several auxin influx transporters, including AUX1-5, are the most extensively studied(Yu et al, 2015; Zhao et al, 2015; Wang et al, 2019; Ye et al, 2021). However, based on the available results, we were unable to confirm whether the proteins encoded by these three genes are involved in the 1,9-D release.
In addition, four main classes of transporter proteins are currently thought to be involved in the release of metabolites from roots to the rhizosphere: the ABC protein family, the MATE family, the major facilitator superfamily (MFS), and the aluminum-activated malate transporter (ALMT) family (Weston et al, 2012; Zhang M X et al, 2022). ABC transporters are widely distributed among higher plants and are involved in the secretion of secondary metabolites from roots (Buer et al, 2007; Badri et al, 2008; Zhang M X et al, 2022). The release of BNIs from sorghum roots is suppressed by the ABC transporter inhibitor vanadate(Coskun et al, 2017a; Zhang M X et al, 2022),supporting a potential role for ABC transporters in BNI release. MATE proteins are responsible for the transport of citrates, benzoxazoles, artemisinins, inulin, phenols, alkaloids, and flavonoids, which depends on counteriongradients, typically the trans-plasma-membrane gradient of H+. Methyl 3-(4-hydroxyphenyl) propionate and sakuranetin released from sorghum roots are a phenolic and a flavonoid, respectively, and are potential transport substrates for MATE. The activity of plasma membrane H+-ATPases clearly affects the release of BNIs from sorghum roots, underscoring the importance of proton coupling in BNI transport (Zhao and Dixon, 2009; Doshi et al, 2017). Taken together, the available data suggest that MATE transporters are involved in the release of BNIs (Sivaguru et al, 2013; Doshi et al, 2017). Members of the MFS protein family can function as antiporters, co-transporters, and uniporters. In rice, one MFS protein, TOM1, mediates the release of deoxymugineic and avenic acid from roots, suggesting a possible role of MFS proteins during BNI release as well (Nozoye et al,2011; Weston et al, 2012). The ALMT protein is implicated in aluminum resistance by mediating the release of malate ions from roots to the rhizosphere, but whether it can also serve as a transporter of 1,9-D needs to be ascertained yet (Ryan et al, 2011; Weston et al, 2012).
Interestingly, among the enriched and up-regulated genes in the C7/C3 group, there were nine genes encoding ABC proteins, including(),(),(),(),(),(),(),(), and(), four genes encoding MATE proteins, such as,,, and, and three MFS protein-related genes,,, and(Fig. 7-A to -C; Table S11). By contrast, only one ABC transporter, STAR2 (LOC_ Os05g02750), and one MATE transporter, LOC_ Os08g39370, were identified and seen as up-regulated in the N7/C7 group, and the results were corroborated by qRT-PCR analysis (Fig. S4). Whether the transporters responsible for 1,9-D release from different rice varieties are different from those induced by NH4+is an interesting question and a worthy subject for further research.
To further pinpoint which transporters play key roles in 1,9-D release, we suggest the following: (i) ectopic expression of the transporters in frog oocytes and determination of 1,9-D secretion; (ii) application of CRISPR/Cas9 and gene overexpression motifs to construct knockout and overexpressed transgenic rice lines and measurement of 1,9-D release in these lines to clarify the roles of various gene candidates in regulating 1,9-D secretion.
In summary, we here have performed a multi-omic analysis to identify candidate genes involved in 1,9-D biosynthesis and transport. We found that the regulation of 1,9-D release in different rice genotypes (WYJ7 and WYJ3) and in response to NH4+treatments may not employ the same biosynthetic pathways and transporters. In future work, we hope to identify linkage regions associated with 1,9-D release by genome-wide association analysis (GWAS) of rice varieties with different 1,9-D-secretion abilities or by detection of QTLs for 1,9-D release from WYJ7/WYJ3 populations. Joint analysis of candidate genes obtained by multi- omic analysis and linkage regions identified by GWAS and QTLs, as well as genetic transformation of callus tissue, should assist in definitively and efficiently pinpointing the genes implicated in 1,9-D biosynthesis and those governing its release from roots.
methods
Rice materials and growth conditions
Two rice varieties, WYJ7 (high 1,9-D release) and WYJ3 (no 1,9-D release), were used. Seeds were surface-sterilized with 3% H2O2for 1 h, rinsed with distilled water for three times, and germinated on 0.5 mmol/L CaCl2at 30 ºC for 2 d. Subsequently, germinated seeds were transferred into 1/2 modified Kimura solution for a 4-day pretreatment. The solution composition was as follows: 0.5 mmol/L NH4NO3, 0.18 mmol/L KH2PO4, 0.54 mmol/L MgSO4·7H2O, 0.18 mmol/L KCl, 0.36 mmol/L CaCl2, 0.2 μmol/L CuSO4·5H2O, 0.5 μmol/L MnCl2·4H2O, 0.4 μmol/L ZnSO4·7H2O, 3 μmol/L H3BO3, 1 μmol/L (NH4)6Mo7O24·4H2O, and 20 μmol/L Na2EDTA-Fe. The solution pH was 5.8. Seedlings were cultivated in a growth chamber, with a 16 h/8 h and 30 ºC/28 ºC light/dark cycle, 400 μmol/(m2·s) light intensity, and a relative humidity of 65%. The root exudates were collected from 6-week-old seedling roots(Zhang et al, 2019; Lu et al, 2022).
Root exudate collection and 1,9-D determination
Thirty 6-week-old seedlings (= 4) from each replicate were rinsed three times consecutively with deionized water prior to use. The washed seedlings were then transferred to a tall, light-proof glass beaker and the roots were gently immersed in 1 L of 1.0 mmol/L NH4Cl (NH4+treatment) or 1.0 mmol/L CaCl2(control, CK) and assayed for 1,9-D release. The detailed collection method and test procedure were described in our previous study(Zhang et al, 2019).
Metabolite identification using GC-MS and data analysis
Accurately weighed samples (60 mg) and two small steel balls were added to a 1.5-mL tube. A methanol and water mixture (at a ratio of 1:1, containing 4 μg/mL of l-2-chlorophenylalanine) of 600 μL was added and placed at -40 ºC for 2 min. Subsequently, the samples were ground at 60 Hz for 2 min and then extracted by ultrasonic for 30 min in an ice-water bath. Chloroform (50 μL) was added to the samples and vortexed for another 2 min. The samples were extracted by ultrasonic for 30 min in an ice-water bath, then placed at -40 ºC for 30 min and centrifuged at 4 ºC (13 000 r/min) for 10 min. Supernatant (150 μL) was collected in a glass vial and dried in a freeze- concentration centrifugal dryer. Next, 80 μL of 15 mg/mL methoxylamine hydrochloride in pyridine was added and the mixture was incubated at 37 ºC for 60 min. Then, 50 μL of bis(trimethylsilyl)-trifluoroacetamide (with 1% trimethylchlorosilane) and 20 μL-hexane were added into the mixture, and derivatized at 70 ºC for 60 min. The samples were further placed at ambient temperature for 30 min prior to GC-MS analysis.
The samples were analyzed on an Agilent 7890B gas chromatography system coupled to an Agilent 5977B MSD system (Agilent Technologies Inc., CA, USA). An AHP-5MS fused-silica capillary column (30 m × 0.25 mm × 0.25 μm, Agilent J & W Scientific, Folsom, CA, USA) was utilized to separate the derivatives. The original data were imported into the MS-DIAL software and further characterized based on the LUG database (Untarget database of GC-MS from Lumingbio, www.lumingbio.com). Subsequently, principal component analysis, OPLS-DA, and partial least squares-discriminant analysis were utilized to observe and distinguish the overall distribution among the samples and the stability of the analysis. To prevent overfitting, 7-fold cross-validation and 200 response permutation testing were used to evaluate the quality of the model. Variable importance of projection (VIP) values, obtained from the OPLS-DA model and two-tailed Student’s-test, were further used to rank the overall contribution of each variable and to verify whether the differences in metabolites between groups were significant. DEMs were selected with VIP > 1.0 and< 0.05.
Metabolite identification using LC-MS and data analysis
Root samples (60 mg) and two small steel balls were added to the tube. A methanol and water mixture (at a ratio of 7:3, containing 4 μg/mLof l-2-chlorophenylalanine) of 600 μL was added to each sample and placed at -40 ºC for 2 min. Then, the samples were ground at 60 Hz for 2 min, and extracted by ultrasonic for 30 min in an ice-water bath before being placed overnight at -40 ºC. Samples were centrifuged at 4 ºC (13 000 r/min) for 10 min, and 150-μL supernatants were collected from each tube using crystal syringes. The samples were filtered through 0.22-μm microfilters and transferred to LC vials prior to determination.
The samples were analyzed using an ACQUITY UPLC I-Class system (Waters corporation, Milford, USA) coupled to a VION IMS QTOF mass spectrometer (Waters Corporation, Milford, USA). The original LC-MS data were processed using Progenesis QI V2.3 software (Nonlinear, Dynamics, Newcastle, UK) and qualitatively analyzed by using The Human Metabolome Database (HMDB), Lipidmaps (V2.3), Metlin, EMDB, PMDB, and self-built databases. Subsequently, data quality control used the same metrics as were used in GS-MS. The valid DEMs were selected with VIP > 1.0 and< 0.05.
RNA extraction and RNA-sequencing
Total RNA samples were extracted using a mirVana™ miRNA Isolation Kit (QIAGEN, Darmstadt, Germany) following the manufacturer’s protocol. The quality of RNA was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The detailed methods of synthesizing and purifying the first-strand and double-strand cDNA and the sample library construction were as described in our previous studies (Sun et al,2017, 2020). Subsequently, the library products were sequenced by Illumina NovaSeqTM6000 (Illumina, San Diego, USA).
DEGs were analyzed using the NOISeq method. The valid DEGs were chosen with a threshold of< 0.05 and a fold change value > 2. The Short Time-series Expression Miner program (version 1.3.11) was used to construct a hierarchical cluster, which was used to exhibit the expression patterns of genes in different samples. GO enrichment and KEGG pathway enrichment analyses of DEGs were performed using R software, as also described in detail in our previous studies (Sun et al,2017, 2020).
Integrated analysis of DSMs and DEGs
The Pearson’s correlation test was performed to screen the correlations between the DSMs and DEGs (< 0.05) (https:// cloud.oebiotech.cn/task/). Subsequently, DEGs and DSMs were further mapped to the KEGG database and their common pathway information was thus obtained.
qRT-PCR analysis
Total RNA was extracted using the FastPure Universal Plant Total RNA Isolation Kit (RC411; Vazyme Biotech Co., Ltd, Nanjing, China). Total RNA (500 ng) was used for reverse transcription by using a HiScript 1st-Strand cDNA Synthesis Kit (R111; Vazyme Biotech Co., Ltd, Nanjing, China), and then cDNA was diluted (20 times) prior to qRT-PCR analysis. The qRT-PCR program was as described (Di et al, 2018). The primers are listed in Table S12.
Statistical analysis
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant Nos. 32030099 and 32072670), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA28020301), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (Grant No. 2023326), and the Enterprise Cooperation Projects of China (Grant No. Am20210407RD). We thank Shanghai OE Biotech Co., Ltd and Shanghai Luming Biological Technology Co., Ltd, Shanghai, China for transcriptome and metabolome analyses.
SupplementaL data
The following materials are available in the online version of this article at http://www.sciencedirect.com/journal/rice-science; http://www.ricescience.org.
Fig. S1. Response sorting tests (200) of orthogonal partial least squares-discriminant analysis (OPLS-DA) model of gas chromatography-mass spectrometer.
Fig. S2. Response sorting tests (200) of orthogonal partial least squares-discriminant analysis (OPLS-DA) model of liquid chromatography-mass spectrometer.
Fig. S3. KEGG analysis of up-regulated and down-regulated differentially synthesized metabolites (DSMs, top20) in NH4+-WYJ7 vs CK-WYJ7 and CK-WYJ7 vs CK-WYJ3.
Fig. S4. Relative transcription of genes encoding 1,9-decanediol transporters in CK-WYJ3, CK-WYJ7 and NH4+-WYJ7.
Table S1. Differentially synthesized metabolites identified by gas chromatography-mass spectrometer.
Table S2. KEGG analysis of NH4+-WYJ7 vs CK-WYJ7 and CK-WYJ7 vs CK-WYJ3 using gas chromatography-mass spectrometer.
呼吸困难是急性期中重度COPD患者常见的临床症状,是患者病情复发住院的主要诱发因素之一[5]。而肺康复治疗的主要观察指标是患者的呼吸困难症状是否获得有效改善[6]。通过呼吸及功能锻炼能较快经鼻深吸气,且能让肋间外肌、膈肌等吸气肌参与到整个吸气过程中,使进入肺中的气量尽可能多,能有效防止气道过早的陷闭,吸气后再缓慢地缩唇呼气,延长呼气时间,以减少呼气末期肺内潴留的二氧化碳含量,利于气体交换,从而有效改善患者呼吸困难症状[7]。
Table S3. Differentially synthesized metabolites identified by liquid chromatography-mass spectrometer.
Table S4. KEGG analysis of NH4+-WYJ7 vs CK-WYJ7 and CK-WYJ7 vs CK-WYJ3 using liquid chromatography-mass spectrometer.
Table S5. Statistics of RNA-seq data.
Table S6. Total differentially expressed unigenes identified in NH4+-WYJ7 vs CK-WYJ7 and CK-WYJ7 vs CK-WYJ3.
Table S7. GO enrichment analysis in NH4+-WYJ7 vs CK- WYJ7 and CK-WYJ7 vs CK-WYJ3.
Table S8. KEGG enrichment analysis in NH4+-WYJ7 vs CK-WYJ7 and CK-WYJ7 vs CK-WYJ3.
Table S9. Co-upregulated genes in NH4+-WYJ7 vs CK-WYJ7 and CK-WYJ7 vs CK-WYJ3.
Table S10. Differentially synthesized metabolites correlated to differentially expressed unigenes in NH4+-WYJ7 vs CK- WYJ7 and CK-WYJ7 vs CK-WYJ3.
Table S11. Up-regulated genes involved in transporter activity in NH4+-WYJ7 vs CK-WYJ7 and CK-WYJ7 vs CK-WYJ3.
Table S12. Primers used in this study.
Badri D V, Loyola-Vargas V M, Broeckling C D, De-la-Peña C, Jasinski M, Santelia D, Martinoia E, Sumner L W, Banta L M, Stermitz F, Vivanco J M. 2008. Altered profile of secondary metabolites in the root exudates ofATP-binding cassette transporter mutants., 146(2): 762–771.
Buer C S, Muday G K, Djordjevic M A. 2007. Flavonoids are differentially taken up and transported long distances in Arabidopsis., 145(2): 478–490.
Chen Z C, Yamaji N, Motoyama R, Nagamura Y, Ma J F. 2012. Up-regulation of a magnesium transporter geneis required for conferring aluminum tolerance in rice., 159(4): 1624–1633.
Cheng H, Kong W P, Tang T X, Ren K L, Zhang K L, Wei H X, Lin T. 2022. Identification of key gene networks controlling soluble sugar and organic acid metabolism during oriental melon fruit development by integrated analysis of metabolic and transcriptomic analyses., 13: 830517.
Coskun D, Britto D T, Shi W M, Kronzucker H J. 2017a. How plant root exudates shape the nitrogen cycle., 22(8): 661–673.
Coskun D, Britto D T, Shi W M, Kronzucker H J. 2017b. Nitrogen transformations in modern agriculture and the role of biological nitrification inhibition., 3: 17074.
Czarnota M A, Paul R N, Weston L A, Duke S O. 2003. Anatomy of sorgoleone-secreting root hairs ofspecies., 164(6): 861–866.
Di D W, Sun L, Zhang X N, Li G J, Kronzucker H J, Shi W M. 2018. Involvement of auxin in the regulation of ammonium tolerance in rice (L.)., 432(1): 373–387.
Doshi R, McGrath A P, Piñeros M, Szewczyk P, Garza D M, Kochian L V, Chang G. 2017. Functional characterization and discovery of modulators of SbMATE, the agronomically important aluminium tolerance transporter from., 7: 17996.
Elrys A S, Desoky E S M, Abo El-Maati M F, Elnahal A S, Abdo A I, Raza S, Zhou J B. 2019. Can secondary metabolites extracted fromseeds suppress ammonia oxidizers to increase nitrogen use efficiency and reduce nitrate contamination in potato tubers?, 185: 109689.
Elrys A S, Uwiragiye Y, Zhang Y H, Abdel-Fattah M K, Chen Z X, Zhang H M, Meng L, Wang J, Zhu T B, Cheng Y, Zhang J B, Cai Z C, Chang S X, Müller C. 2023. Expanding agroforestry can increase nitrate retention and mitigate the global impact of a leaky nitrogen cycle in croplands., 4(1): 109–121.
Fliegmann J, Schüler G, Boland W, Ebel J, Mithöfer A. 2003. The role of octadecanoids and functional mimics in soybean defense responses., 384(3): 437–446.
Fu M Y, Yang X, Zheng J R, Wang L, Yang X Y, Tu Y, Ye J B, Zhang W W, Liao Y L, Cheng S Y, Xu F. 2021. Unraveling the regulatory mechanism of color diversity inpetals by integrative transcriptome and metabolome analysis., 12: 685136.
Kaur-Bhambra J, Wardak D L R, Prosser J I, Gubry-Rangin C. 2022. Revisiting plant biological nitrification inhibition efficiencyusing multiple archaeal and bacterial ammonia-oxidising cultures., 58(3): 241–249.
Kronzucker H J, Siddiqi M Y, Glass A D M. 1997. Conifer root discrimination against soil nitrate and the ecology of forest succession., 385: 59–61.
Kronzucker H J, Siddiqi M Y, Glass A D, Kirk G J. 1999. Nitrate- ammonium synergism in rice: A subcellular flux analysis., 119(3): 1041–1046.
Li T L, Wang Z G, Wang C X, Huang J Y, Feng Y F, Shen W S, Zhou M, Yang L Z. 2022. Ammonia volatilization mitigation in crop farming: A review of fertilizer amendment technologies and mechanisms., 303: 134944.
Liu X Y, Sheng J, Curtiss R. 2011. Fatty acid production in genetically modified cyanobacteria., 108(17): 6899–6904.
Lu X F, Vora H, Khosla C. 2008. Overproduction of free fatty acids in.: Implications for biodiesel production., 10(6): 333–339.
Lu Y F, Zhang X N, Jiang J F, Kronzucker H J, Shen W S, Shi W M. 2019. Effects of the biological nitrification inhibitor 1,9-decanediol on nitrification and ammonia oxidizers in three agricultural soils., 129: 48–59.
Lu Y F, Zhang X N, Ma M K, Zu W J, Kronzucker H J, Shi W M. 2022. Syringic acid from rice as a biological nitrification and urease inhibitor and its synergism with 1,9-decanediol., 58(3): 277–289.
Ma M K, Lu Y F, Di D W, Kronzucker H J, Dong G Q, Shi W M. 2023. The nitrification inhibitor 1,9-decanediol from rice roots promotes root growth inthrough involvement of ABA and PIN2-mediated auxin signaling., 280: 153891.
Min J, Shi W M. 2018. Nitrogen discharge pathways in vegetable production as non-point sources of pollution and measures to control it., 613/614: 123–130.
Min J, Sun H J, Wang Y, Pan Y F, Kronzucker H J, Zhao D Q, Shi W M. 2021. Mechanical side-deep fertilization mitigates ammonia volatilization and nitrogen runoff and increases profitability in rice production independent of fertilizer type and split ratio., 316: 128370.
Nardi P, Laanbroek H J, Nicol G W, Renella G, Cardinale M, Pietramellara G, Weckwerth W, Trinchera A, Ghatak A, Nannipieri P. 2020. Biological nitrification inhibition in the rhizosphere: Determining interactions and impact on microbially mediated processes and potential applications., 44(6): 874–908.
Nozoye T, Nagasaka S, Kobayashi T, Takahashi M, Sato Y, Sato Y, Uozumi N, Nakanishi H, Nishizawa N K. 2011. Phytosiderophoreefflux transporters are crucial for iron acquisition in graminaceous plants., 286(7): 5446–5454.
O’Sullivan C A, Fillery I R P, Roper M M, Richards R A. 2016. Identification of several wheat landraces with biological nitrification inhibition capacity., 404(1): 61–74.
Otaka J, Subbarao G V, Ono H, Yoshihashi T. 2022. Biological nitrification inhibition in maize: Isolation and identification of hydrophobic inhibitors from root exudates., 58(3): 251–264.
Reiser S, Somerville C. 1997. Isolation of mutants ofdeficient in wax ester synthesis and complementation of one mutation with a gene encoding a fatty acyl coenzyme A reductase., 179(9): 2969–2975.
Ryan P R, Tyerman S D, Sasaki T, Furuichi T, Yamamoto Y, Zhang W H, Delhaize E. 2011. The identification of aluminium-resistance genes provides opportunities for enhancing crop production on acid soils., 62(1): 9–20.
Sivaguru M, Liu J P, Kochian L V. 2013. Targeted expression of SbMATE in the root distal transition zone is responsible for sorghum aluminum resistance., 76(2): 297–307.
Souza E F C, Rosen C J, Venterea R T. 2021. Co-application of DMPSA and NBPT with urea mitigates both nitrous oxide emissions and nitrate leaching during irrigated potato production., 284: 117124.
Steen E J, Kang Y S, Bokinsky G, Hu Z H, Schirmer A, McClure A, Del Cardayre S B, Keasling J D. 2010. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass., 463: 559–562.
Streubel J, Pesce C, Hutin M, Koebnik R, Boch J, Szurek B. 2013. Five phylogenetically close ricegenes confer TAL effector-mediated susceptibility topv.., 200(3): 808–819.
Subbarao G V, Ito O, Sahrawat K L, Berry W L, Nakahara K, Ishikawa T, Watanabe T, Suenaga K, Rondon M, Rao I M. 2006. Scope and strategies for regulation of nitrification in agricultural systems: Challenges and opportunities., 25(4): 303–335.
Subbarao G V, Tomohiro B, Masahiro K, Osamu I, Samejima H, Wang H Y, Pearse S J, Gopalakrishnan S, Nakahara K, Zakir Hossain A K M, Tsujimoto H, Berry W L. 2007. Can biological nitrification inhibition (BNI) genes from perennial() combat nitrification in wheat farming?, 299(1): 55–64.
Subbarao G V, Nakahara K, Ishikawa T, Yoshihashi T, Ito O, Ono H, Ohnishi-Kameyama M, Yoshida M, Kawano N, Berry W L. 2008. Free fatty acids from the pasture grassand one of their methyl esters as inhibitors of nitrification., 313(1): 89–99.
Subbarao G V, Nakahara K, Hurtado M P, Ono H, Moreta D E, Salcedo A F, Yoshihashi A T, Ishikawa T, Ishitani M, Ohnishi- Kameyama M, Yoshida M, Rondon M, Rao I M, Lascano C E, Berry W L, Ito O. 2009. Evidence for biological nitrification inhibition inpastures., 106: 17302–17307.
Subbarao G V, Nakahara K, Ishikawa T, Ono H, Yoshida M, Yoshihashi T, Zhu Y Y, Zakir H A K M, Deshpande S P, Hash C T, Sahrawat K L. 2013a. Biological nitrification inhibition (BNI) activity in sorghum and its characterization., 366(1): 243–259.
Subbarao G V, Rao I M, Nakahara K, Sahrawat K L, Ando Y, Kawashima T. 2013b. Potential for biological nitrification inhibition to reduce nitrification and N2O emissions in pasture crop- livestock systems., 7: 322–332.
Subbarao G V, Sahrawat K L, Nakahara K, Rao I M, Ishitani M, Hash C T, Kishii M, Bonnett D G, Berry W L, Lata J C. 2013c. A paradigm shift towards low-nitrifying production systems: The role of biological nitrification inhibition (BNI)., 112(2): 297–316.
Subbarao G V, Yoshihashi T, Worthington M, Nakahara K, Ando Y, Sahrawat K L, Rao I M, Lata J C, Kishii M, Braun H J. 2015. Suppression of soil nitrification by plants., 233: 155–164.
Subbarao G V, Kishii M, Bozal-Leorri A, Ortiz-Monasterio I, Gao X, Ibba M I, Karwat H, Gonzalez-Moro M B, Gonzalez-Murua C, Yoshihashi T, Tobita S, Kommerell V, Braun H J, Iwanaga M. 2021. Enlisting wild grass genes to combat nitrification in wheat farming: A nature-based solution., 118(35): e2106595118.
Sun L, Lu Y F, Yu F W, Kronzucker H J, Shi W M. 2016. Biological nitrification inhibition by rice root exudates and its relationship with nitrogen-use efficiency., 212(3): 646–656.
Sun L, Di D W, Li G J, Kronzucker H J, Shi W M. 2017. Spatio- temporal dynamics in global rice gene expression (L.) in response to high ammonium stress., 212: 94–104.
Sun L, Di D W, Li G J, Li Y L, Kronzucker H J, Shi W M. 2020. Transcriptome analysis of rice (L.) in response to ammonium resupply reveals the involvement of phytohormone signaling and the transcription factor OsJAZ9 in reprogramming of nitrogen uptake and metabolism., 246/247: 153137.
Vioque J, Kolattukudy P E. 1997. Resolution and purification of an aldehyde-generating and an alcohol-generating fatty acyl-CoA reductase from pea leaves (L.)., 340(1): 64–72.
Wang M, Qiao J Y, Yu C L, Chen H, Sun C D, Huang L Z, Li C Y, Geisler M, Qian Q, Jiang D A, Qi Y H. 2019. The auxin influx carrier, OsAUX3, regulates rice root development and responses to aluminium stress., 42(4): 1125–1138.
Wang R, Ren C X, Dong S, Chen C, Xian B, Wu Q H, Wang J, Pei J, Chen J. 2021. Integrated metabolomics and transcriptome analysis of flavonoid biosynthesis in safflower (L.) with different colors., 12: 712038.
Wang X, Bai J H, Wang C, Xie T, Wang W, Wang D W, Zhang G L. 2023. Two newly-identified biological nitrification inhibitors in: Synthetic pathways and influencing mechanisms., 454: 140172.
Weston L A, Ryan P R, Watt M. 2012. Mechanisms for cellular transport and release of allelochemicals from plant roots into the rhizosphere., 63(9): 3445–3454.
Woodward E E, Hladik M L, Kolpin D W. 2016. Nitrapyrin in streams: The first study documenting off-field transport of a nitrogen stabilizer compound., 3(11): 387–392.
Ye R G, Wu Y R, Gao Z Y, Chen H, Jia L X, Li D M, Li X G, Qian Q, Qi Y H. 2021. Primary root and root hair development regulation byand its participation in the phosphate starvation response., 63(8): 1555–1567.
Yu C L, Sun C D, Shen C J, Wang S K, Liu F, Liu Y, Chen Y L, Li C Y, Qian Q, Aryal B, Geisler M, Jiang D A, Qi Y H. 2015. The auxin transporter, OsAUX1, is involved in primary root and root hair elongation and in Cd stress responses in rice (L.)., 83(5): 818–830.
Zakir H A K M, Subbarao G V, Pearse S J, Gopalakrishnan S, Ito O, Ishikawa T, Kawano N, Nakahara K, Yoshihashi T, Ono H, Yoshida M. 2008. Detection, isolation and characterization of a root-exuded compound, methyl 3-(4-hydroxyphenyl) propionate, responsible for biological nitrification inhibition by sorghum ()., 180(2): 442–451.
Zhang M, Fan C H, Li Q L, Li B, Zhu Y Y, Xiong Z Q. 2015. A 2-yr field assessment of the effects of chemical and biological nitrification inhibitors on nitrous oxide emissions and nitrogen use efficiency in an intensively managed vegetable cropping system., 201: 43–50.
Zhang M X, Zeng H Q, Afzal M R, Gao X, Li Y X, Subbarao G V, Zhu Y Y. 2022. BNI-release mechanisms in plant root systems: Current status of understanding., 58(3): 225–233.
Zhang X N, Lu Y F, Yang T, Kronzucker H J, Shi W M. 2019. Factors influencing the release of the biological nitrification inhibitor 1,9-decanediol from rice (L.) roots., 436(1/2): 253–265.
Zhang Y T, Yang L W, Yang J J, Hu H L, Wei G Q, Cui J B, Xu J. 2022. Transcriptome and metabolome analyses reveal differences in terpenoid and flavonoid biosynthesis inneedles across different seasons., 13: 862746.
Zhao H M, Ma T F, Wang X, Deng Y T, Ma H L, Zhang R S, Zhao J. 2015. OsAUX1 controls lateral root initiation in rice (L.)., 38(11): 2208–2222.
Zhao J, Dixon R A. 2009. MATE transporters facilitate vacuolar uptake of epicatechin 3ʹ--glucoside for proanthocyanidin biosynthesis inand., 21(8): 2323–2340.
Zheng Y N, Li L L, Liu Q, Yang J M, Wang X W, Liu W, Xu X, Liu H, Zhao G, Xian M. 2012. Optimization of fatty alcohol biosynthesis pathway for selectively enhanced production of C12/14 and C16/18 fatty alcohols in engineered., 11: 65.
1 June 2023;
11 August 2023
Shi Weiming (wmshi@issas.ac.cn)
Copyright © 2024, China National Rice Research Institute. Hosting by Elsevier B V
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/)
Peer review under responsibility of China National Rice Research Institute
http://doi.org/10.1016/j.rsci.2023.09.002
(Managing Editor: Wu Yawen)
猜你喜欢
家庭医药(2023年2期)2023-05-30
保健与生活(2021年17期)2021-09-17
中华养生保健(2020年2期)2020-11-16
云南医药(2019年3期)2019-07-25
心肺血管病杂志(2019年4期)2019-06-27
国际呼吸杂志(2019年5期)2019-03-30
现代养生·上半月(2017年10期)2017-10-12
中国当代医药(2015年17期)2015-03-01
中国医药科学(2015年15期)2015-02-27
中华皮肤科杂志(2014年3期)2014-12-19
- Rice Science的其它文章
- Rice Variety Classification Based on Optimized Near-Infrared Spectral Classification Model
- Effects of Nitrogen-Regulating Gene AreA on Growth,Pathogenicity,and Fumonisin Synthesis of Fusarium proliferatum
- Rice Husk at a Glance:From Agro-Industrial to Modern Applications
- Smart Farming for Sustainable Rice Production:An Insight into Application,Challenge, and Future Prospect
- A β-Carotene Ketolase Gene NfcrtO from Subaerial Cyanobacteria Confers Drought Tolerance in Rice
- OsbZIP01 Affects Plant Growth and Development by Regulating OsSD1 in Rice