
ldentification of immune cell-related prognostic genes characterized by a distinct microenvironment in hepatocellular carcinoma
2024-03-07 04:29MengTingLiKaiFengZhengYiErQiu
Meng-Ting Li,Kai-Feng Zheng,Yi-Er Qiu
Abstract BACKGROUND The development and progression of hepatocellular carcinoma (HCC) have been reported to be associated with immune-related genes and the tumor microenvironment.Nevertheless,there are not enough prognostic biomarkers and models available for clinical use.Based on seven prognostic genes,this study calculated overall survival in patients with HCC using a prognostic survival model and revealed the immune status of the tumor microenvironment (TME).AIM To develop a novel immune cell-related prognostic model of HCC and depict the basic profile of the immune response in HCC.METHODS We obtained clinical information and gene expression data of HCC from The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium(ICGC) datasets.TCGA and ICGC datasets were used for screening prognostic genes along with developing and validating a seven-gene prognostic survival model by weighted gene coexpression network analysis and least absolute shrinkage and selection operator regression with Cox regression.The relative analysis of tumor mutation burden (TMB),TME cell infiltration,immune checkpoints,immune therapy,and functional pathways was also performed based on prognostic genes.RESULTS Seven prognostic genes were identified for signature construction.Survival receiver operating characteristic curve analysis showed the good performance of survival prediction.TMB could be regarded as an independent factor in HCC survival prediction.There was a significant difference in stromal score,immune score,and estimate score between the high-risk and low-risk groups stratified based on the risk score derived from the seven-gene prognostic model.Several immune checkpoints,including VTCN1 and TNFSF9,were found to be associated with the seven prognostic genes and risk score.Different combinations of checkpoint blockade targeting inhibitory CTLA4 and PD1 receptors and potential chemotherapy drugs hold great promise for specific HCC therapies.Potential pathways,such as cell cycle regulation and metabolism of some amino acids,were also identified and analyzed.CONCLUSION The novel seven-gene (CYTH3, ENG, HTRA3, PDZD4, SAMD14, PGF,and PLN) prognostic model showed high predictive efficiency.The TMB analysis based on the seven genes could depict the basic profile of the immune response in HCC,which might be worthy of clinical application.
Key Words: Hepatocellular carcinoma;Prognostic model;Weighted gene coexpression network analysis;Microenvironment;Chemotherapy
lNTRODUCTlON
Hepatocellular carcinoma (HCC) is a common cancer that has the highest prevalence among liver cancer subtypes.It is well known that the mortality of HCC has increased gradually and is likely to break through millions of cases[1].Despite rapid technological and medical advances in new tests and treatments for HCC patients,their overall five-year survival rate is less than 5%[2].In addition,since HCC has no specific clinical manifestations,patients might have been diagnosed in the late stages.In this regard,it is necessary to explore novel prognostic biomarkers and risk models to help forecast overall survival (OS) of HCC patients with greater accuracy.
The immune system and immune cells are both considered to be essential for developing various cancers,including HCC[3].A previous study reported that an imbalance in immune regulation affected the cancer microenvironment and contributed to tumor progression and metastasis[4].The altered crosstalk between tumor cells and the immune system repre-sents a new hallmark in the diagnosis and treatment of HCC.For instance,CD8+CXCR5+T cells migrate in response to supernatants from primary HCC (HCC-SN) cells and cause patients with HCC to have a worse prognosis[5].The activation of tumor-associated macrophages could contribute to increasing the number of HCC cellsviadestabilization of adherens junctions[6].Likewise,increasing the number and activity of mature dendritic cells could improve the prognosis of HCC patients[7].To date,the specific status of different cell subtypes and programmed cell death have been explored in the tumor microenvironment (TME) of HCC[8,9].However,few researchers have focused on exploring the specific association or mechanism between immune infiltration and liver cancer through the genes of all immune cells.Moreover,potential clinical applications along with the mechanism of different types of immune cell-related genes regulating the TME for HCC progression remain unclear and need further research.
In this research,we first identified immune cell-related genes (ICRGs) through weighted gene coexpression network analysis (WGCNA) in The Cancer Genome Atlas (TCGA) datasets to explore the potential prognostic immune-related genes that were most enriched and correlated with HCC.After least absolute shrinkage and selection operator regression(LASSO) regression with Cox regression analysis,we identified seven ICRGs to establish and validate a well-worked prognostic model in the TCGA and International Cancer Genome Consortium (ICGC) datasets.Meanwhile,a series of bioinformatic analyses,including protein-protein interaction,immune checkpoint analysis,immune checkpoint blockade,chemotherapy drug analysis,gene set variation analysis (GSVA),and gene set enrichment analysis,were also performed to determine how all immune cell-related genes may impact HCC's TME in this study.The results of this scenario also helped determine potential biomarkers that might be novel therapeutic targets in HCC.Our study also identified biomarkers that help determine the best therapeutic strategy for each patient.
MATERlALS AND METHODS
Datasets
The clinical information of HCC patients and corresponding cancer tissue RNA-seq data were identified and retrieved from the TCGA (https://portal.gdc.cancer.gov/repository) for model construction and ICGC (https://dcc.icgc.org/projects/LIRI-JP/) database for validation.There were 374 HCC tissues and 50 normal tissues in the TCGA dataset,and 265 tumor samples in the ICGC dataset.
ICRGs
First,we identified the hub immune-cell-related genes through weighted gene coexpression network analysis (WGCNA).A co-expression network of ICRGs was built by using the R package “WGCNA v1.68”.After a topological overlapping matrix was performed to detect modules,405 genes in black module were chosen and regarded as ICRGs.Next,we identified 338 differentially expressed ICRGs with the screening standard set asP< 0.05.In addition,String (Protein-Protein Interaction Networks,V: 10.5) database (https://string-db.org/) analysis on the 338 differentially expressed ICRGs was performed to obtain the interactions between proteins-targeted genes.Then,51 hub ICRGs were filtered out by performing univariate Cox regression analysis (P< 0.05).
Construction and validation of a prognostic model
A penalized shrunken regression method named LASSO was conducted on the β-coefficients to minimize potential overfitting.Based on the results of the Cox regression analysis,seven prognostic ICRGs were identified and used to construct a prognostic model.We calculated and obtained each patient’s risk score according to the formula below: Risk score=esum (each gene’s expression × corresponding coefficient).Then,a model for immune-cell-related prognostic indicators was developed as follows: Risk score=∑I=1 N (Explg*Coef).The median risk score could be used to divide HCC samples into low-risk (< median) and high-risk (≥ median) groups.
Prognostic and clinical implications
OS probability analysis and the time-dependent receiver operating characteristic (ROC) curve analysis based on the risk model and seven prognostic ICRGs were performed using the R package “survival”,“survminer”,“survivalROC”,or“timeROC”.Using the R package “survival”,we performed multivariate and univariate prognostic analyses based on the TCGA and ICGC datasets,respectively.In addition,the area under the curve (AUC) values of the seven prognostic ICRGs,the risk model,and various clinical factors for predicting the survival at 1,3,and 5 years were calculated.
The available clinical factors,like age,gender,tumor grade,and tumor stage,were compared to assess the clinical implication of the model and depicted using the R package “ggpubr”.In order to improve the possibility of usage in clinical practice,a nomogram was constructed and the calibration curves were plotted based on the seven prognostic ICRGs with the help of the R package “rms”.
Analysis of mutational load and TME
We assessed the status of TMB and somatic mutations in HCC in the two risk groups with the help of the R package“maftools”.The Kaplan-Meier curves were also obtained after HCC samples were divided into high-TMB or low-TMB groups.
Next,we further depicted the TME of HCC in the risk model by assessing the immune-related functions of immune cells through single-sample gene set enrichment analysis (ssGSEA) using the R package “gsva”.For assessing immune infiltration subtype between the high-and low-risk groups,we performed two-way ANOVA.The relationship between tumor stemness and risk score was assessed by Spearman correlation analysis.ESTIMATE algorithm was used to explore the levels of cell infiltration by calculating the immune and stromal scores.
Immune checkpoint analysis and immune checkpoint blockade
Immune checkpoint (ICP) analysis was conducted to determine and visualise the correlations among genes related to ICPs,the seven ICRGs,and risk scores by using the R packages “limma”,“BiocManager”,“reshape2”,and “ggplot2”.The immunophenoscore (IPS) of HCC samples was collected from the cancer-immune group atlas (https://tcia.at/home).The higher the IPS score,the higher the positive correlation with increased immunogenicity.The tumor immune dysfunction and exclusion (TIDE) algorithm (http://tide.dfci.harvard.edu) was used to predict the response to immune checkpoint blockade (ICB) treatment in the two risk groups.
Chemotherapy drug analysis
To predict the half-maximal inhibitory concentration (IC50) of chemotherapeutic agents for each patient in the two risk groups,the R package “pRRophetic” (https://github.com/paulgeeleher/pRRophetic) was used.However,based on the risk model,a great number of chemotherapeutic drug were found.In order to conduct further research on HCC,we selected four commonly used drugs that had been used in clinical trials.
Meanwhile,data of NCI-60 human cancer cell lines from a publicly available dataset from CellMiner project page (https://discover.nci.nih.gov/cellminer) were also downloaded to determine the connection between the seven ICRGs and drug sensitivity.
Functional enrichment analysis
We determined the potential functional pathways that might play an essential role in HCC progression and TME regulation by GSVA and gene set enrichment analysis (GSEA) with the help of the R packages “BiocManager”,“limma”,“GSEABase”,and “GSEV”.
RESULTS
WGCNA and identification of hub modules
To screen the hub genes related to the TME characteristic index in HCC patients,the RNA-seq data and relevant clinical data of samples (normal:tumor=50:374) were downloaded from TCGA.The samples were used to identify outliers with the help of hierarchical clustering analysis and no samples were found to be outliers (Figure 1A).Next,we set the β-value to 20 to ensure that the network was scale-free (Figure 1B).After the method of dynamic cutting was implemented,separate gene co-expression modules were divided.Similar modules with a difference < 25% were combined (Figure 1C and D).Modules of similar sizes were screened and merged using the height parameter of 0.25,deep split parameter of 4,and minModuleSize parameter of 60.As a result of WGCNA,we were able to obtain six modules for further research(Figure 1E).In order to ascertain the hub modules,a Pearson test (P< 0.05) was employed to compute the correlation between the characteristic genes of said module and immune-infiltrated cells.Then,based on the criteria of correlation >0.1 andP< 0.05,we calculated the numbers of immune cells in each module to ascertain the appropriate immune-related module.The 405 genes in the black module were selected for the subsequent research because they had 13 immune cells that met the criteria,compared with 10 in red,8 in lightcyan,8 in tan,11 in purple,and 12 in gray.
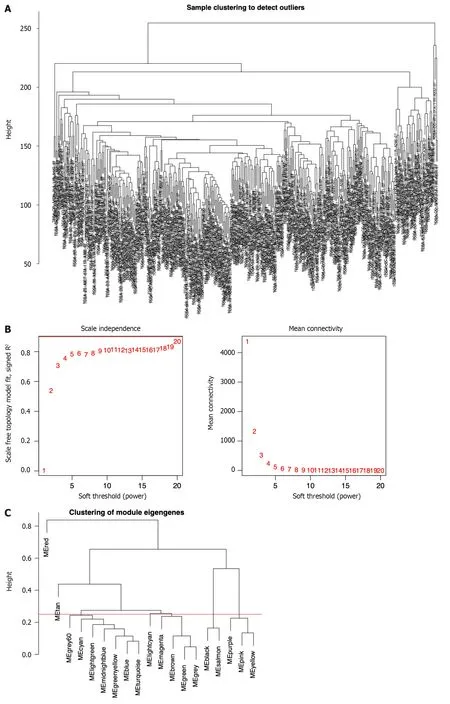
Figure 1 ldentification of immune-related differential genes by weighted gene coexpression network analysis in the The Cancer Genome Atlas database. A: Sample clustering analysis to detect outliers;B: Analysis of the scale-free fit index of the soft threshold power (β);C: The module merge threshold indicated by a horizontal line;D: Hierarchical cluster tree showing the results of modules in weighted gene coexpression network analysis;E: Heatmap analysis showing the associations between the module characteristic genes and immune cell infiltration.
Identification of ICRGs
We next identified ICRGs from the 405 genes in the black module.A total of 338 ICRGs were identified and expressed differentially between normal and HCC tissues (Figure 2A).A protein-protein interaction network was constructed to show that the genes could be roughly divided into four categories: Laminin family members,collagen family members,ADAMTS family members,and the Notch signaling network (Figure 2B).Then,we used univariate Cox regression to identify the hub ICRGs that played an essential role in model construction,and 51 hub ICRGs were obtained (Figure 2C).

Figure 2 ldentification of immune cell-related genes by array screening. A: Heatmap of the identified immune cell-related genes (ICRGs) between the tumor group and the normal group.Blue: Low expression level;red: High expression level;B: Protein-protein interaction network of the genes (cutoff=0.9);C:Univariate Cox regression analysis of ICRGs.
Establishment of a prognostic signature based on seven prognostic ICRGs
Seven prognostic ICRGs (CYTH3,ENG,HTRA3,PDZD4,SAMD14,PGF,andPLN) were screened out to develop a prognostic signature through the utilization of LASSO Cox regression analysis (Figure 3A and B).According to the formula of risk score=0.369451059941312 * CYTH3+-0.287762938289728 * ENG+0.221566259099006 * HTRA3+-1.00820639333083 * PDZD4+0.542684310219352 * SAMD14+0.526196082946206 * PGF+-0.393399578080273 * PLN,the risk score of each patient was calculated.All patients could be divided into high-and low-risk groups based on the seven prognostic ICRGs in the two datasets (Figure 3C and F).Using the median cutoff value of risk scores for patents in the TCGA and ICGC datasets,two groups with high-risk and low-risk scores were also classified (Figure 3D and G).Moreover,as supported by the higher density of red dots observed in the high-risk area within both cohorts,the patients who belonged to the high-risk group might have a worse outcome (Figure 3E and H).
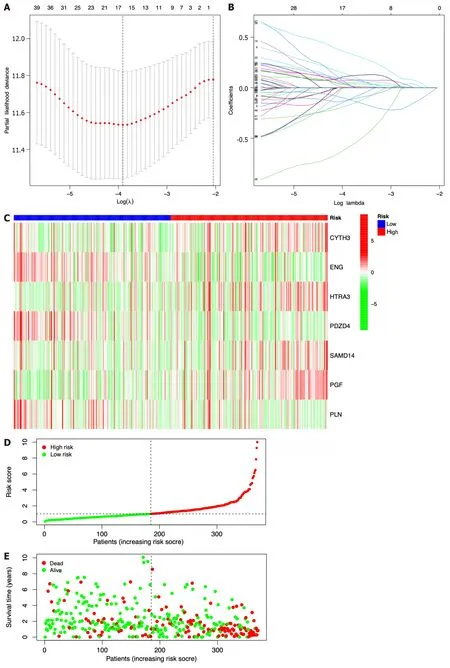
Figure 3 Construction of a prognostic model based on seven identified immune cell-related genes using The Cancer Genome Atlas and lnternational Cancer Genome Consortium datasets. A: Lasso Cox regression analysis of 51 hub immune cell-related genes (ICRGs) after univariate Cox regression;B: Partial likelihood deviance for different numbers of variables.In The Cancer Genome Atlas (TCGA) dataset,the seven-gene prognostic signature was evaluated;C: Heatmap of related genes;D: Distribution of overall survival (OS) status;E: The median value and distribution of the risk scores.In the International Cancer Genome Consortium dataset,the seven-gene prognostic signature was evaluated;F: Heatmap of related genes;G: Distribution of OS status;H: The median value and distribution of the risk scores.
Prognostic implication of the seven-ICRG prognostic model
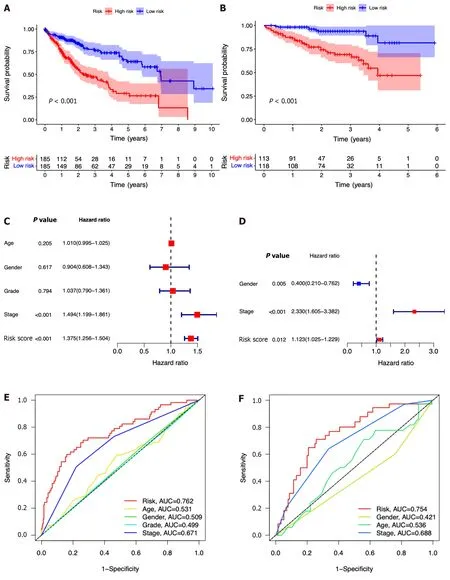
In comparison to those with lower risk scores,patients with higher risk scores exhibited a notably diminished OS in the TCGA dataset,which was further verified in the ICGC dataset (Figure 4A and B).Simultaneously,the potential significance of the risk score as a variable factor in prognostic prediction was also confirmed through univariate Cox regression analysis.In this study,we conducted multivariate survival analyses in both TCGA and ICGC datasets to demonstrate the efficacy of the model.The results revealed a hazard ratio (HR) of 1.375 [95% confidence interval (CI):1.256-1.504;P< 0.001] in TCGA and an HR of 1.123 (95%CI: 1.025-1.229;P=0.012) in ICGC (Figure 4C and D).The AUC values demonstrated that the seven-ICRG prognostic model had the highest predictable value in TCGA (AUC=0.762)and ICGC (AUC=0.754) compared to other traditional features such as tumor stage,age,sex,and tumor grade (Figure 4E and F).The AUC values obtained from the time-dependent ROC analysis were 0.762,0.748,and 0.757 at time points of 1,3,and 5 years in TCGA and 0.794,0.778,and 0.776 in ICGC,respectively (Figure 4G and H).
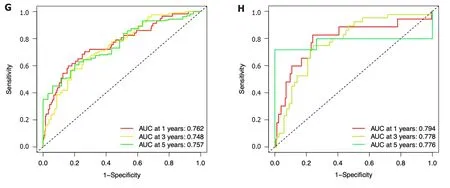
Figure 4 The seven-gene prognostic signature demonstrates high predictive power for overall survival in patients with hepatocellular carcinoma. A: Kaplan-Meier curves for overall survival (OS) of patients in the high-and low-risk groups in The Cancer Genome Atlas (TCGA) cohort;B: Kaplan-Meier curves for OS of patients in the high-and low-risk groups in the immune cell-related gene (ICRG) cohort;C: Prognosis-related variables screened by multivariate Cox regression were analyzed in the TCGA cohort;D Prognosis-related variables screened by multivariate Cox regression were analyzed in the ICGC cohort;E: Area under the time-dependent ROC curves (AUC) for different clinical features in the TCGA cohort;F: AUC for different clinical features in the ICGC cohort;G: AUC of the prognostic model for survival at different time points in the TCGA cohort ;H: AUC of the prognostic model for survival at different time points in the ICGC cohort.
Survival probability associated with the seven ICRGs and nomogram construction
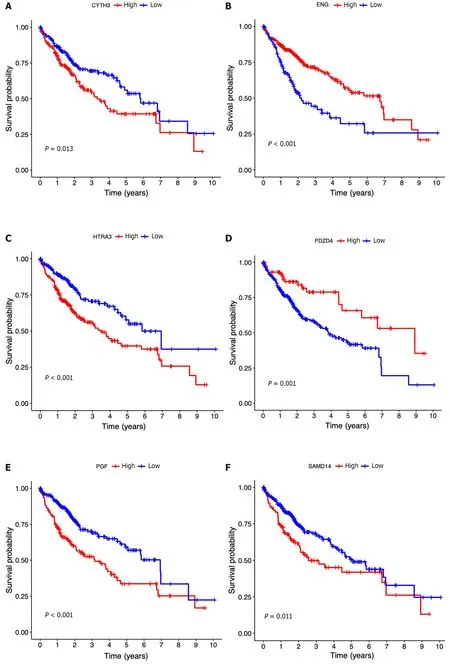
Then,the patients diagnosed with HCC were categorized into separate subgroups based on the seven ICRGs to conduct a more comprehensive investigation into the prognostic significance of individual genes within the signature.We found that the survival probabilities associated withCYTH3,HTRA3,PGF,andSAMD14were significantly lower in the highrisk group compared to the low-risk group,whileENG,PDZD4,andPLNshowed the opposite trend (Figure 5A-G).
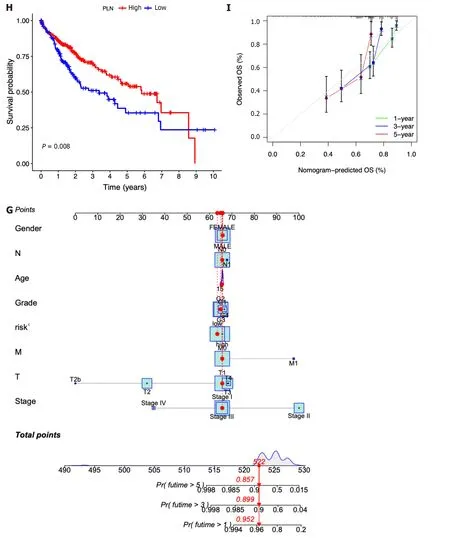
Figure 5 Clinical implications of the seven-lCRG prognostic model. A-G: Kaplan-Meier analyses for survival by CYTH3 (A),ENG (B),HTRA3 (C),PDZD4 (D),PGF (E),PLN (F),and SAMD14 (G);H: Nomogram for predicting hepatocellular carcinoma patient survival;I: Calibration plots applied for predicting the 1-,3-,and 5-year overall survival in the The Cancer Genome Atlas cohort.
In addition,according to the fundamental prognostic variables from the multivariate Cox regression analysis,we developed a nomogram to accurately forecast the OS rates for HCC patients over a span of 1 year,3 years,and 5 years.Initially,patients have the opportunity to accrue points by summing the values of all variables within the nomogram.Subsequently,the cumulative points for each patient can be computed to derive the 1-,3-,and 5-year OS rates,employing a vertical line extending from the prognostic factor axis to the points axis (Figure 5H).Figure 5I exhibits a notable level of accuracy of the nomogram in predicting the OS of HCC patients at the time of 1 year,3 years,and 5 years.
Clinical traits of the seven-ICRG prognostic model
In each dataset,we evaluated the clinical implications of clinical factors such as age,sex,tumor grade,and tumor stage as explanatory variables in the seven-ICRG prognostic model.We noticed that patients with high tumor grade and stage III-IV were more likely to have a higher risk score in TCGA,and a similar trend occurred in stage III-IV in ICGC datasets(Figure 6C,D,and G).However,no significant differences were observed in age or sex (Figure 6A,B,E,and F).
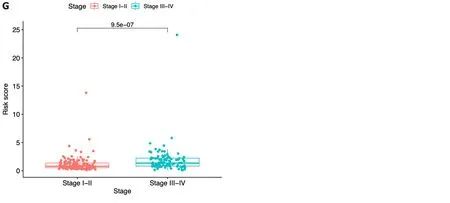
Figure 6 Risk score according to various clinical characteristics. A-D: Risk score according to age (A),gender (B),tumor grade (C),and tumor stage (D)based on The Cancer Genome Atlas dataset;E-G: Risk score according to age (E),gender (F),and tumor stage (G) based on the immune cell-related gene dataset.
Genomic features of the seven-ICRG prognostic model
According to the results of somatic mutation analysis,in the low-risk group,mutations in CTNNB1 (26%),TTN (21%),TP53 (17%),and MUC16 (13%) were highly enriched.Concurrently,the high-risk group exhibited a significant enrichment of mutations in TP53 (38%),TTN (26%),CTNNB1 (25%),and MUC16 (16%).Missense mutation was the main type of genetic variation (Figure 7A and B).As the risk score increased,we also observed increased tumor mutation burden (TMB) in tumor samples (Figure 7C).The data presented in Figure 7D indicated that patients belonging to the low-TMB group exhibited a significantly elevated probability of survival,while patients in the high-TMB group displayed a notably diminished probability of survival (P< 0.001).When combining risk and TMB classification,the lowest survival probability occurred in patients in the high-risk+high-TMB group,while the highest survival probability occurred in the low-risk+low-TMB group (P< 0.001,Figure 7E).

Figure 7 Relationship among the seven-gene prognostic model,genomic features,and tumor mutation burden. A: List of the most frequently altered genes in the low-risk group;B List of the most frequently altered genes in the high-risk group;C: Scatter plot of the relationship between the risk score and tumor mutation burden (TMB);D: Kaplan-Meier analysis of survival probability for the high-and low-risk groups;E: Kaplan-Meier analysis of survival probability for the high-TMB+high-risk,high-TMB+low-risk,low-TMB+high-risk,and low-TMB+low-risk groups.H-TMB: High-TMB;L-TMB: Low-TMB.
Tumor microenvironment analysis
In this study,we investigated the potential correlation between different immune cell types and the risk scores of patients in the TCGA dataset.Based on the outcomes of the analyses conducted using seven software applications (XCELL,TIMER,QUANTISEQ,MCPCOUNTER,EPIC,CIBERSORT-ABS,and CIBERSORT),our observations were that CD8+T cells,mast cells,etc.were related with the decrease in patient risk,while CD4+T cells,B cells,M0 macrophages,and regulatory T cells (Tregs) were associated with the increase in patient risk scores (Figure 8A).Subsequently,we performed ssGSEA to assess and compare the ssGSEA scores across distinct risk groups,thereby elucidating the disparities in immune infiltration between the high-and low-risk cohorts.It was observed that macrophages exhibited a more pronounced association with the high-risk group,whereas B cells,CD8+T cells,dendritic cells,mast cells,neutrophils,natural killer cells,plasmacytoid dendritic cells,T helper cells,and tumor infiltrating lymphocytes were found to be linked with the low-risk group (Figure 8B,P< 0.05).Then,we conducted a comparative analysis of the immune functions in the two groups in order to investigate the potential involvement of immune pathways in their differentiation.Our research revealed that the high-risk group exhibited greater activity of major histocompatibility complex class I,while antigen presenting cell coinhibition,cytolytic activity,human leukocyte antigen,inflammation promotion,and type I and type II IFN responses in the low-risk group exhibited higher levels of activity (Figure 8C,P<0.05).
Next,we introduced four distinct immune infiltration types,namely,wound healing (C1),interferon gamma (IFN-γ)dominant (C2),inflammatory (C3),and lymphocyte depleted (C4),in order to ascertain their association with the signature.The findings revealed a strong correlation between C1 and high-risk patients,whereas C2,C3,and C4 were more likely to manifest in individuals with low-risk scores (Figure 8D).
Then,the RNA stemness score (RNAs) and DNA methylation pattern (DNAss) were used to evaluate tumor stemness.As presented in Figure 8E-F,no significant correlation was observed between the DNAss and risk score,while a more positive association was identified between the RNAss and risk score (r=0.24,P< 0.05).In addition,the risk score was significantly associated with the stromal score,immune score,and estimate score of patients with HCC in the low-risk group,implying a more essential role of the TME in the low-risk group than in the high-risk group (Figure 8G).
ICP analysis and ICB therapy
The expression association between 47 immune checkpoints and the risk score or the seven ICRGs was also studied.The results showed that the levels of most immune checkpoints,such as VTCN1,TNFSF9,TNFSF4,TNFSF18,TNFSF15,TNFRSF9,TNFRSF4,and TNFRSF18,were positively related to the risk score.However,TMIGD2,PDCD1LG2,and IDO2 were negatively correlated with the risk score (Figure 9A).IPS analysis was also conducted for the prediction of patient responsiveness to CTLA-4 and PD-1.The results showed that the IPS,IPS-CTLA4,IPS-PD1,and IPS-PD1-CTLA4 scores were higher in the low-risk group (P< 0.05,Figure 9B-E).
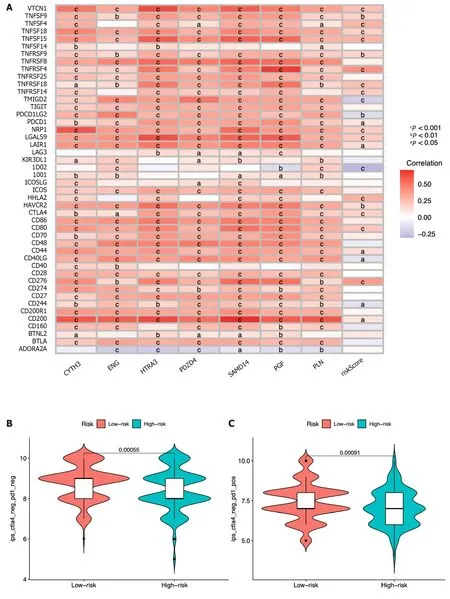
Figure 9 lmmune-check point analysis of the seven identified immune cell-related genes and immune checkpoint blockade. A: Heatmap of association among 47 immune checkpoints,risk score,and the seven identified immune cell-related genes;B-E: Association between the tumor microenvironment and risk score;F: Violin plot showing the relationship of tumor immune dysfunction and risk score between the low-and high-risk groups.cP < 0.001,bP < 0.01,aP <0.05.
In order to conduct a comprehensive assessment of the efficacy the risk score as a prognostic indicator for patients undergoing ICB therapy,a TIDE analysis was conducted on HCC patients from the TCGA dataset.It was observed that patients classified within the high-risk group exhibited lower TIDE scores,indicating a potential heightened responsiveness to ICB therapy in comparison to the low-risk group (Figure 9F).
Chemotherapy drug analysis
By using the R package “pRRophetic”,the IC50analysis of four relatively frequently used chemotherapeutic drugs for HCC was conducted in the two groups.Axitinib,dasatinib,and erlotinib were observed to have significant differences in estimated IC50between the two groups.In addition,we noticed that patients in the high-risk group had higher IC50values,implying that patients in the low-risk group were more sensitive to these three drugs (Figure 10A-C).Meanwhile,the IC50value of gemcitabine was found to be higher in the low-risk group compared to the high-risk group,indicating a greater likelihood of gemcitabine sensitivity among patients in the high-risk group (Figure 10D).
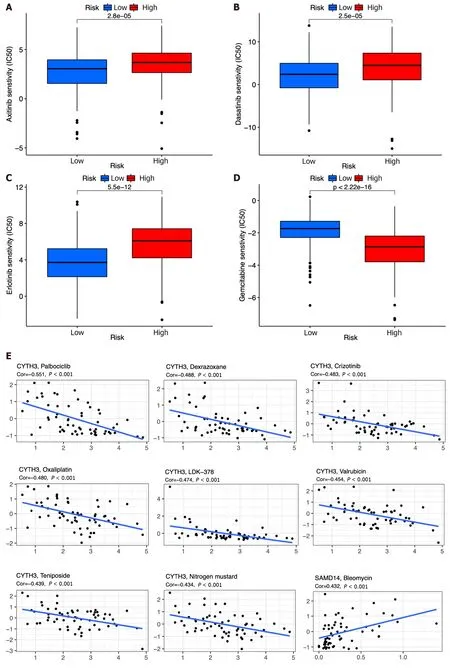
Figure 10 Chemotherapy drug analysis between the high-and low-risk groups in the The Cancer Genome Atlas cohort. A-D: Different sensitivities to axitinib (A),dasatinib (B),erlotinib (C),and gemcitabine (D) between the high-and low-risk groups;E: Scatter plots of the top 16 correlation analyses between immune cell-related genes and drug sensitivity.
We also explored the top 16 correlation analyses between prognostic genes and drug sensitivity according to NCI-60.Figure 10E demonstrates that patients withSAMD14expression were sensitive to bleomycin.However,patients withCYTH3expression were insensitive to palbociclib,dexrazoxane,crizotinib,oxaliplatin,LDK-37,valrubicin,teniposide,nitrogen mustard,LEE-011,DAUNORUBICIN,raloxifene,etoposide,epirubicin,and daunorubicin.In addition,patients with expression ofPDZD4were insensitive to palbociclib (Figure 10E).
Identification of biological pathways associated with the seven ICRGs and risk score
The predictive ability of the seven ICRGs and the risk score can be attributed to their key roles in tumorigenesis and metastasis.Thus,we further explored the richness differences by GSVA on the seven ICRGs and risk score.As GSEA enrichment depicted in Figure 11A,we observed that several pathways that are correlated with tumorigenesis and T cells were positively related to the risk score,including WNT_BETA_CATENIN_SIGNALING,TGF_BETA_SIGNALING,PI3K_AKT_MTOR_SIGNALING,MYC_TARGETS,MTORC1_SIGNALING,MITOTIC_SPINDLE,GLYCOLYSIS,G2M_CHECKPOINT,E2F_TARGETS,and DNA_REPAIR.Some pathways were negatively related to the risk score,such as XENOBIOTIC_METABOLISM,PEROXISOME,and KRAS_SIGNALING.
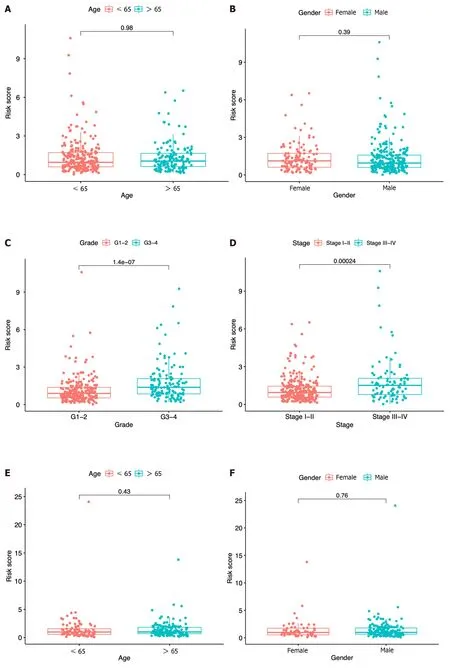

Figure 11 Bioinformatic analysis of the risk model. A: Gene set variation analysis of the seven identified immune cell-related prognostic genes and the risk score;B: Gene set enrichment analysis of biological functions and pathways between the high-and low-risk groups.cP < 0.001,bP < 0.01,aP < 0.05.
Meanwhile,GSEA method was employed to investigate the potential involvement of signaling pathways in HCC patients of the two groups.We selected the ten pathways with the highest enrichment scores,and the findings indicated that PYRIMIDINE_METABOLISM,BASE_EXCISION_REPAIR,RNA_DEGRADATION,NUCLEOTIDE_ EXCISION_REPAIR,and CELL_CYCLE were more related to the high-risk group,while TRYPTOPHAN_METABOLISM,VALINE_LEUCINE_AND_ISOLEUCINE_DEGRADATION,FATTY_ACID_METABOLISM,GLYCINE_ SERINE_AND_THREONINE_METABOLISM,and BUTANOATE_METABOLISM were more related to the low-risk group(Figure 11B).
DlSCUSSlON
In this study,we identified seven prognostic ICRGs from all the immune cell-related genes in the TCGA database and established and validated a well-worked prognostic model for HCC patients based on the TCGA and ICGC datasets.To the best of our knowledge,this was the first time that the prognostic genes of HCC were screened from all the immune cell-related genes by WGCNA.To date,plenty of research has explored the associations between specific subtypes of immune cells and HCC to construct a prognostic model based on them.For example,a four-gene signature and nomogram related to macrophages were constructed,and the immune landscape with abnormal infiltration of macrophages was depicted as well[10].The infiltration of CD8+T cells and the expression of MMP9 were closely related to the OS of HCC patients[11].However,these studies,which only explored the infiltration of a single subtype of immune cells,ignored the fact that immune cells could cooperate or antagonize each other to promote the progression and metastasis of different cancers,including HCC.Innate immune cells,such as dendritic cells,Kupffer cells,and natural killer T cells,interact with the T-cell response to regulate the TME of HCC[12].Macrophage-lymphocyte interactions were also observed to be a key element of cancer-related inflammation[13].In addition,cross-talk between Tregs and mast cells was allowed by a positive feedback system that functioned through TGF-β1 and IL-9 in gastric cancer development[14].This evidence suggests that we should also explore the mechanism and TME of HCC from more immune cell subtypes rather than just a single subtype of immune cells.
With this conjecture,we screened seven prognostic genes in the black module,namely,CYTH3,ENG,HTRA3,PDZD4,SAMD14,PGF,andPLN,from the entire immune cell gene pool through WGCNA,LASSO regression,and Cox regression analysis.Interestingly,what we found proved the correctness of our conjecture.Here,we observed that the prognostic model based on the seven ICRGs had a good predictive ability in the TCGA and ICGC datasets,which was verified by the finding that patients with higher risk scores had a shorter OS.The AUC values of the various clinical features indicated that the risk score exhibited the highest level of predictability.Through time-dependent ROC analysis,the AUC values were determined to be 0.762,0.748,and 0.757 at 1,3,and 5 years based on the TCGA dataset,and they were 0.794,0.778,and 0.776 at 1,3 and 5 years based on the ICGC dataset,respectively,which all indicated that the signature worked well in the prediction of HCC patients’ survival.Then,we noticed that the seven ICRGs in the black module had a strong correlation with the T-cell population,which consisted of CD8+T cells,resting memory CD4+T cells,activated memory CD4+T cells,follicular helper T cells,and regulatory T cells (Tregs) in HCC[15].Thus,we mainly focused on the subtypes of T cells here.After a thorough literature search,it was found that the expression ofENG,HTRA3,PDZD4,PGF,andPLNwas reported to be associated with HCC,whileHTRA3,PDZD4,andPLNwere only reported to be included in the prognostic model of HCC[16,17].High endoglin (ENG) expression in endothelial cells might activate HCC cell adhesion and endothelial functions[18].ENG is also regarded as a mediator that stimulates T-cell activation,proliferation,and Th1 cytokine secretion to promote T-cell-mediated cytolysis in cancer treatment[19].PGF(placental growth factor) produced by cancer-associated fibroblasts facilitates neoangiogenesis in HCC[20].PGF can be secreted by the Th17 subset of helper T cells to promote angiogenesis and thus participate in Th17 differentiation in inflammation[21].For CYTH3 and PDZD4,there have been no reports on their association with either HCC or any subtype of T cells until now.

Next,the genomic features of the seven-ICRG prognostic model were explored.Wanget al[22] found thatTP53andCTNNB1are two of the most commonly mutated genes in Chinese HCC patients.In our research,we found that CTNNB1(26%) was most enriched in the low-risk group,while TP53 (38%) was the most enriched.CTNNB1 activation promoted immune escape by impairing T-cell activity in HCC[23].Somatic mutations in CTNNB1 were found to antagonize increasing levels of T-cell cytotoxicity or immunological shifts toward cytotoxic CD8+T cells,leading to the failure of chemotherapy for HCC[24].TP53,encoded by the most commonly mutated cancer driver gene,might cooperate with EGF domain-specific O-linked N-acetylglucosamine transferase (EOGT) to reduce the proportion of CD8+T cells and impair regulatory T-cell differentiation in HCC[25].Here,we noticed that the lowest survival probability occurred in patients in the high-risk and high-TMB group,which indicated that that TMB has the potential to serve as a standalone variable in survival analysis,owing to its capacity to modulate immune status.
Then,the TME of HCC was analyzed based on the risk model.CD8+T cells were more related to a decrease in patient risk,while CD4+T cells and Tregs were associated with higher risk scores.It is well known that elevated levels of cytotoxic CD8+T cells are associated with stronger antitumor effects,and low levels of CD8+T cells are correlated with poor HCC outcomes[26].The presence of CD4+T cells promotes the proliferation and activation of CD8+T cells to avoid CD8+T-cell exhaustion.CD4+T cells are indispensable for the secondary expansion and memory of CD8+T lymphocytes[27].In fact,CD4+T cells often become exhausted as HCC progresses.Furthermore,we conducted ssGSEA and found that CD8+T cells and cytolytic activity were enriched in the low-risk group.No significant difference was observed between the two risk groups in CD4+T cells,which suggested that the collapse of CD8+T cells might contribute to insufficient CD4+T cells.In addition,a further detailed analysis of the immune infiltration of HCC was performed to identify the role of the risk score in immune infiltration.The results showed that the enrichment of C1(wound healing) mainly occurred in patients belonging to the high-risk group in the analysis of the TME.As previously reported,the wound-healing response could participate in promoting the development of HCC[28].T cells coordinate with other proinflammatory cytokines and chemokines to participate in hepatic fibrosis,which is regarded as a woundhealing response in HCC[29].In addition,significant differences were detected between the two risk groups with regard to stromal score,immune score,estimate score,and RNAss,which again emphasized that the seven ICRG-related TME might facilitate the development of HCC and that RNAss could be involved.
A comparison between the risk score and ICPs or ICB therapy was conducted to identify beneficial targets for immunotherapy and evaluate the efficacy of immunotherapy.Immune checkpoints,such as VTCN1,TNFSF9,TNFSF4,TNFSF18,TNFSF15,TNFRSF9,TNFRSF4,and TNFRSF18,were positively related to the risk score,which suggests that targeted and specific treatments should be carried out for the two risk groups.Moreover,the IPS,IPS-CTLA4,IPS-PD1,and IPS-PD1-CTLA4 immune checkpoint scores were higher in the low-risk group,suggesting that patients in the low-risk group might have a better response to immunotherapy.PD-1 and CTLA-4,checkpoint receptors that can be targeted to relieve the exhaustion of CD8+T cells,were enriched in the low-risk group[30].PD1 inhibitors might directly act on CD8+T-cell cells to enhance the antitumor effect in this risk group.
In recent studies,immune checkpoint inhibitors (ICIs) plus tyrosine kinase inhibitors (TKIs),which are antiangiogenic drugs,seemed more effective in antitumor therapy,especially in reversing the immunosuppressive profile of the TME and improving the efficacy,OS,and safety profile[31].We must be more cautious in the utilization of ICIs for HCC treatment,given the escalating incidence of HCC cases attributed to "metabolic" and "metabolic+alcohol" etiologies,as well as the intricate interplay between liver metabolism and immune system regulation[32,33].In addition,Tregs are the most prevalent suppressor cells in the TME and express immune checkpoints such as PD-1 and CTLA-4,showing a potential therapeutic role of targeting Tregs in HCC treatment[34].In our research,we observed an association between Tregs and patients at higher risk,suggesting that PD-1 and CTLA-4 might have the potential to alleviate patients by specifically targeting Tregs.Furthermore,the utilization of transarterial chemoembolization (TACE) has the potential to stimulate the generation of antigen-specific CD4+and CD8+T cells in patients with HCC,thus rendering the combination of TACE and ICI treatment highly promising in terms of its application prospects[35].Radiogenomics research associated with TME heterogeneity has also made progress in recent years.Wanget al[36] established a CT-derived radiomics signature based on seven hub genes and revealed infiltration of CD4+T cells,plasma cells,and macrophages among different risk groups.It is noteworthy that an Austrian team had devised an ART score for assessing the potential efficacy of repeated TACE treatments in patients,although this score had not yet undergone clinical validation[37].These results suggest that additional clinical validation is needed for the exploration of the TME and ICIs in our research.
HCC exhibits significant resistance to various chemotherapy agents,whether administered as monotherapy or in combination.Therefore,it is imperative to select suitable chemotherapy medications that have obtained approval from the Food and Drug Administration for patients at varying levels of risk.In our investigation,it was observed that patients classified in the high-risk category exhibited heightened sensitivity to gemcitabine,whereas axitinib,dasatinib,and erlotinib may potentially yield more favorable outcomes in low-risk patients.Then,the data from a total of 60 distinct cell lines was scrutinized in order to identify additional chemotherapy agents that could augment the efficacy of genetargeted drugs while mitigating the risk of drug resistance.For instance,increased expression ofSAMD14gene was observed to render cancer cells more susceptible to bleomycin.Based on the aforementioned findings,it is possible that future advancements in precision strategies could unveil novel directions for drug treatment in HCC.
In the final analysis,we hoped to identify some representative functional pathways involved in the immune-related progression of HCC,especially those on T-cell regulation.Several pathways,such as the Wnt/β-catenin pathway and the TGF-β signaling pathway,have been reported to correlate with T-cell regulation in the TME of HCC.Inhibition of the Wnt/β-catenin pathway has been reported to boost the infiltration and IFN-γ production of TIL CD8+T cells and reduce the number of Tregs[38].Inhibition of TGF-β promoted the activity of CD8+T cells and reduced the action of CD4+Treg cells by interrupting the differentiation of M2-type macrophages in HCC[39].Tregs were induced under stimulation with lactate,viathe PI3K/Akt/mTOR signaling pathway,to establish immunosuppression in HCC[40].Meanwhile,GSEA between the high-and low-risk groups showed that cell cycle regulation (PYRIMIDINE_METABOLISM,BASE_EXCISION_REPAIR,RNA_DEGRADATION,NUCLEOTIDE_EXCISION_REPAIR and CELL_CYCLE) might be an important pathway in the TME of HCC.Although T-cell proliferation rarely occurred in infiltration analysis of HCC in this study,other studies have observed that Tregs were elevated and proliferated to fight against the antitumor immunity of HCC patients[41,42].The metabolism of some amino acids (tryptophan,alanine,leucine,glycine,threonine,etc.) was related to the low-risk group.The metabolism of various amino acids in shaping tumor immunity and therapeutic efficacy in patients with cancer has long been investigated[43].For example,alanine deprivation delays naive and memory T-cell activation to impact the TME[44].By adding tryptophan,IDO(+) MΦ-suppressed T-cell responses could be reversed in the TME of HCC[45].T cells showed a defective response to antigen stimulation in SLC7A5-deficient mice because of leucine transportation failure[46].Targeting the metabolism of various amino acids might be worthy of clinical application in the treatment of HCC.
CONCLUSlON
In conclusion,we identified seven prognostic genes that are closely related to the immune-related TME in HCC.A novel prognostic model based on the seven ICRGs was constructed to predict the OS and prognosis of HCC patients.The related potential functional information and TME profile of the seven ICRGs were explored and depicted in HCC as well.The established prognostic model and several potential chemotherapy drugs identified could provide useful insights into the potential clinical treatment of HCC.
ARTlCLE HlGHLlGHTS
Research background
Hepatocellular carcinoma (HCC) is a common cancer that has the highest prevalence among liver cancer subtypes.The overall five-year survival rate of HCC is less than 5%.Thus,it is necessary to explore novel prognostic biomarkers and risk models for this malignancy.
Research motivation
To investigate the relationship between immune cells and HCC,and establish a prognostic model.
Research objectives
To develop a novel immune cell-related prognostic model of HCC and depict the basic profile of the immune response in HCC.
Research methods
Clinical information and gene expression data of HCC were obtained from The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) datasets.TCGA and ICGC datasets were used for screening prognostic genes along with developing and validating a seven-gene prognostic survival model by weighted gene coexpression network analysis and least absolute shrinkage and selection operator regression with Cox regression.Based on the prognostic genes identified,we further performed the relative analysis of tumor mutation burden,tumor microenvironment cell infiltration,immune checkpoints,immune therapy,and functional pathways.
Research results
Seven prognostic genes were identified for signature construction,which showed a good performance for survival prediction.A significant difference was observed in stromal score,immune score,and estimate score between he highrisk and low-risk groups stratified based on the risk score derived from the seven-gene prognostic model..Several immune checkpoints were found to be associated with the seven prognostic genes and risk score.Targeting CTLA4 and PD1 receptors and potential chemotherapy drugs might be helpful for specific HCC therapies.Cell cycle regulation and metabolism of some amino acids were also identified as special signaling pathways.
Research conclusions
A novel seven-gene (CYTH3,ENG,HTRA3,PDZD4,SAMD14,PGF,andPLN) prognostic model was successfully established and showed high predictive efficiency.The basic profile of the immune response in HCC might be worthy of clinical application.
Research perspectives
The novel seven-gene immune-cell related prognostic model might be useful for revealing the basic profile of immune response in HCC.Potential chemotherapy drugs could provide useful insights into the potential clinical treatment of HCC.
FOOTNOTES
Author contributions:Li MT contributed to writing-original draft;Zheng KF contributed to writing-reviewing and editing;Qiu YE contributed to supervision.
lnstitutional review board statement:Our research was conducted based on public datasets,and no human subjects were involved in this research.Thus,there is no need to obtain approval by the institutional review board.
lnformed consent statement:Our research was conducted based on public datasets,and no human subjects were involved in this research.Thus,there is no need to obtain informed consent.
Conflict-of-interest statement:All the authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data sharing statement:The data sets supporting the results of this article are all included in the article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Meng-Ting Li 0000-0001-5894-757X;Yi-Er Qiu 0000-0002-3721-6644.
S-Editor:Liu JH
L-Editor:Wang TQ
P-Editor:Zhang XD
 World Journal of Clinical Oncology2024年2期
World Journal of Clinical Oncology2024年2期
- World Journal of Clinical Oncology的其它文章
- Unlocking the potential-vitamin D in prostate cancer prevention
- Updates on management of gliomas in the molecular age
- Deregulation of interferon-gamma receptor 1 expression and its implications for lung adenocarcinoma progression
- Elucidating the molecular basis of ATP-induced cell death in breast cancer: Construction of a robust prognostic model
- Population-based X-ray gastric cancer screening in Hiroshima prefecture,Japan
- Endoscopic resection for calcifying fibrous tumors of the gastrointestinal tract