
Elucidating the molecular basis of ATP-induced cell death in breast cancer: Construction of a robust prognostic model
2024-03-07 04:29HaoLingZhangSandaiDoblinZhongWenZhangZhiJingSongBabuDineshYasserTabanaDahhamSabbarSaadMowaffaqAdamAhmedAdamYongWangWeiWangHaoLongZhangSenWuRuiZhaoBarakatKhaled
Hao-Ling Zhang,Sandai Doblin,Zhong-Wen Zhang,Zhi-Jing Song,Babu Dinesh,Yasser Tabana,Dahham Sabbar Saad,Mowaffaq Adam Ahmed Adam,Yong Wang,Wei Wang,Hao-Long Zhang,Sen Wu,Rui Zhao,Barakat Khaled
Abstract BACKGROUND Breast cancer is a multifaceted and formidable disease with profound public health implications.Cell demise mechanisms play a pivotal role in breast cancer pathogenesis,with ATP-triggered cell death attracting mounting interest for its unique specificity and potential therapeutic pertinence.AIM To investigate the impact of ATP-induced cell death (AICD) on breast cancer,enhancing our understanding of its mechanism.METHODS The foundational genes orchestrating AICD mechanisms were extracted from the literature,underpinning the establishment of a prognostic model.Simultaneously,a microRNA (miRNA) prognostic model was constructed that mirrored the gene-based prognostic model.Distinctions between high-and low-risk cohorts within mRNA and miRNA characteristic models were scrutinized,with the aim of delineating common influence mechanisms,substantiated through enrichment analysis and immune infiltration assessment.RESULTS The mRNA prognostic model in this study encompassed four specific mRNAs: P2X purinoceptor 4,pannexin 1,caspase 7,and cyclin 2.The miRNA prognostic model integrated four pivotal miRNAs: hsa-miR-615-3p,hsa-miR-519b-3p,hsa-miR-342-3p,and hsa-miR-324-3p.B cells,CD4+T cells,CD8+T cells,endothelial cells,and macrophages exhibited inverse correlations with risk scores across all breast cancer subtypes.Furthermore,Kyoto Encyclopedia of Genes and Genomes analysis revealed that genes differentially expressed in response to mRNA risk scores significantly enriched 25 signaling pathways,while miRNA risk scores significantly enriched 29 signaling pathways,with 16 pathways being jointly enriched.CONCLUSION Of paramount significance,distinct mRNA and miRNA signature models were devised tailored to AICD,both potentially autonomous prognostic factors.This study's elucidation of the molecular underpinnings of AICD in breast cancer enhances the arsenal of potential therapeutic tools,offering an unparalleled window for innovative interventions.Essentially,this paper reveals the hitherto enigmatic link between AICD and breast cancer,potentially leading to revolutionary progress in personalized oncology.
Key Words: ATP-induced cell death;mRNA;miRNA;Prognostic model;Breast cancer
lNTRODUCTlON
ATP-induced cell death (AICD) has emerged as a distinctive mode of cell death triggered by elevated extracellular ATP levels and is closely associated with the progression of various cancer types[1-4].Cell death serves as a critical process in maintaining the normal functionality of tissues and organs,primarily presenting in two distinct forms: apoptosis,and necrosis[5,6].AICD exerts a dual impact on breast cancer: first,ATP acts as a pro-death signal,participating in the regulation of the activation of death pathways;second,cell death itself leads to an escalation in extracellular ATP levels,forming an interconnected regulatory loop[7-11].Manipulation of ATP levels and ATP receptors in the context of breast cancer can alter biological activities such as breast cancer cell proliferation,invasion,and metastasis[12-15].Studies have suggested that AICD is a pivotal mechanism regulated by both genes and microRNAs (miRNAs),contributing to the modulation of breast cancer[16-19].However,the precise nature of interactions requires further research.Hence,in-depth exploration of the mechanisms underlying AICD holds promising potential for its application in breast cancer treatment.Unraveling the implications of AICD in breast cancer therapy could pave the way for novel therapeutic strategies and targeted interventions.
AICD is a multifaceted process,its intricacy being contingent upon the specific cell type and the surrounding microenvironment[20-23].Nonetheless,several overarching mechanisms have been revealed.First,the activation of purinergic receptors,particularly the P2X purinoceptor 7 receptor (P2X7R),has emerged as a pivotal mechanism,triggering a cascade of events culminating in cell death[8,24].Second,a significant mechanism of AICD involves the elevation of intracellular calcium ion concentration[25,26].Moreover,AICD may also be linked to the activation of inflammatory responses[8].Finally,the disruption of mitochondrial function is crucial in AICD,with cytochrome c release intimately connected with the activation of apoptosis signaling pathways[27-29].
Genes and genetic factors encompass nucleotide sequences that encode either a polypeptide chain or functional RNA,thereby supporting fundamental life processes.They serve as repositories of an organism's genetic information,encompassing data pertaining to its race,blood type,reproduction,growth,apoptosis,and other critical processes.On the other hand,miRNAs,which are RNA molecules approximately 21 to 23 nucleotides in length,are widely prevalent in eukaryotic organisms[30].MiRNAs function as key regulators of gene expression,derived from RNA transcribed by DNA but incapable of being translated into proteins (non-coding RNAs).Through specific binding to target mRNA,miRNAs can exert posttranscriptional control over gene expression,playing pivotal roles in the regulation of gene expression,cell cycle,and developmental sequencing in organisms.Notably,in animals,a single miRNA often possesses the capacity to regulate numerous genes simultaneously.
The association between miRNAs and cancer has garnered significant recent interest[31].miRNAs exert a crucial impact on cancer initiation,progression,and therapeutic responses,exhibiting dichotomous effects that either facilitate tumorigenesis,growth,and metastasis,or that impede tumor formation and development[32-35].The functional roles of miRNAs may vary across different cancer types,with certain miRNAs functioning as tumor suppressors to inhibit cancer growth in specific contexts,while others may function as tumor promoters,promoting cancer cell proliferation and metastasis.
The relationship between AICD and miRNA is governed by two distinct mechanisms.First,certain miRNAs have the capacity to modulate the cell response to ATP by targeting essential genes involved in ATP-related signaling pathways or cell death pathways.Conversely,alterations in ATP levels can prompt cells to adjust the expression patterns of miRNAs,thereby influencing cell survival and death.This reciprocal regulatory mechanism may hold particular significance in tumor cells,as it can influence tumor cell proliferation or apoptosis,consequently impacting tumor development and prognosis.Nevertheless,the current body of research concerning AICD-related miRNAs and their corresponding gene regulation in breast cancer remains limited.Although increasing evidence highlights the considerable diagnostic and prognostic value of AICD and miRNA in breast cancer[18,20],the precise regulatory mechanisms and associated biological processes require further exploration and elucidation.
A prognostic model (signature) is a predictive model designed to estimate potential treatment outcomes based on a patient's current health status.Genetic prognostic models aim to forecast disease progression and survival by utilizing a patient's gene expression data,thereby offering more precise and personalized information to guide clinical decisionmaking.The miRNA prognostic model facilitates more accurate prognosis evaluation by healthcare professionals and aids in making informed treatment choices.Gene prognostic models have certain limitations including high-dimensional data,overfitting issues,data consistency concerns,and challenges in biological interpretation.However,the miRNA prognostic model is characterized by the inherent instability of miRNA expression data,data quality problems,miRNAgene interactions,and limitations in study samples.These factors collectively pose challenges for miRNA prognostic models to thoroughly elucidate disease mechanisms.Considering the strengths and weaknesses outlined above,the simultaneous utilization of both gene prognostic models and miRNA prognostic models represents a promising research direction.By harnessing the advantages of both approaches,the prognostic features of cancer can be more effectively explored,providing a more robust tool for personalized treatment and prognosis assessment.
However,no studies to date have investigated the impact of AICD regulatory mechanisms in breast cancer.Therefore,the primary goal of this paper is to look into the potential prognostic importance of AICD genes in breast cancer,as well as the interaction among AICD genes and prognostic gene-associated miRNAs in breast cancer.Initially,a comprehensive literature search was conducted to identify genes associated with AICD,and subsequently,a novel AICD signature with prognostic value was constructed independently of classical clinicopathological parameters.Subsequent functional enrichment and immune infiltration analysis were conducted to further elucidate the role of AICD in regulating fundamental biological processes in breast cancer.The findings of this study show that AICD could be a potential candidate for breast cancer detection and therapeutic intervention,opening up a new research channel and perspective for breast cancer diagnostics and treatment.This discovery holds promise in providing valuable insights for precision treatment and accurate prognosis assessment of breast cancer.
MATERlALS AND METHODS
Literature search of AICD core genes
The literature search aimed to identify core genes associated with AICD using the following keywords: “AICD,” “cell death,” “apoptosis,” “autophagy,” “necrosis,” “death,” and “extracellular ATP.” The search was conducted between March 10,2023 and June 20,2023.Databases including PubMed,Embase,Web of Science,MESH,and Scopus were employed to identify genes related to AICD.The scope of the search was expanded to encompass genes associated with AICD across all diseases,given the limited availability of literature reporting AICD specifically in breast cancer.Publications that did not mention AICD were excluded from the analysis.
Source of datasets
The miRNA and mRNA expression profiles,along with clinical information,were obtained from the UCSC Xena platform,sourced from The Cancer Genome Atlas Breast Cancer (TCGA-BRCA) dataset.To establish and validate the models,the dataset was randomly partitioned into a training set (70%) and an internal validation set (30%).Furthermore,additional miRNA and mRNA expression profile data were obtained,as well as clinical information,from the meatbric database for external validation.During the analysis,samples with missing clinical information were carefully excluded,and samples with a survival period of less than 10 d were also excluded to ensure the reliability and accuracy of the study.
Construction and validation of mRNA- and miRNA-related prognostic models
In this study,R software version 4.1.0 was used for comprehensive data analysis.Initially,the univariate Cox proportional hazards model regression,Least Absolute Shrinkage and Selection Operator (LASSO) regression,and multifactor Cox regression analyses were conducted using the glmnet (version 2.0.18) and survival (version 2.44.1.1) R packages.Univariate Cox regression enabled the assessment of the association between mRNA and miRNA expression levels with overall survival (OS),whereP< 0.05 was considered statistically significant.As miRNAs were inferred from mRNAs,thePvalue threshold for miRNAs was adjusted to < 0.15 to indicate statistical significance.
Following this,LASSO regression analyses were performed on mRNAs and miRNAs that met the specified criteria,further refining the features.Subsequently,the prognostic effect and hazard ratio of the prediction model were evaluated using multivariate Cox regression analysis,and the 95% confidence intervals were calculated.
Using the formula Σ (expmiRNAn × β mirnan),the prognostic risk score was computed by summing the miRNA expression values and their respective coefficients.This risk score facilitated the stratification of samples into high-and low-risk groups.Kaplan-Meier (KM) analysis and bilateral log-rank testing were used with the “survminer” software package to determine the prognostic significance of the risk scores in training,internal validation,whole cohorts,and external validation,with a significance level set atP< 0.05.
Additionally,the “timeROC” package was used to produce time-dependent receiver operating characteristic (ROC)curves to evaluate the prediction model's performance,with the area under the curve (AUC) providing an estimate of the model's accuracy.Overall,all above-mentioned indexes achievedP< 0.05,indicating significant differences.These comprehensive analyses will help validate and assess the efficacy of the constructed mRNA and miRNA prognostic models in predicting survival and risk for patients with breast cancer.
mRNAs corresponded to the prediction of miRNAs and prediction of target mRNAs at miRNA-binding sites
In this study,miRNA-mRNA target prediction was conducted using the miRTarBase database (https://mirtarbase.cuhk.edu.cn/~miRTarBase/miRTarBase_2022/php/index.php) on July 21,2023.Simultaneously,prognostic features of miRNA-targeted mRNA in humans were downloaded from the TargetScan databases (http://www.targetscan.org/vert_80/,accessed on July 23,2023).Subsequently,differential gene expression analysis was performed between the high-and low-risk groups using the “limma” package in R.For the mRNA prognostic model,differential genes were identified based on the criteria of adjustedP< 0.05 and |log2(fold change)| > 1.Conversely,in the miRNA prognostic model,due to the limited number of differential genes,they were defined as genes with an adjustedP< 0.05 and |log2(fold change)|> 0.5.These differential genes will undergo further analysis to evaluate their functional significance and relevance in the prognostic models.
Functional enrichment analysis
In this study,microbial informatics analysis tools (https://www.bioinformatics.com.cn/,accessed on July 24,2023) were employed for conducting gene enrichment analysis,encompassing both Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses.The criterion for determining statistically significant differences was set atP< 0.05,which made it possible identify functional items and pathways that exhibited significant enrichment in the GO and KEGG analyses.
Correlation analysis of immune infiltration in the mRNA prognostic model
This study utilized the Tumor Immune Estimation Resource (http://timer.cistrome.org/,accessed on July 24,2023) to investigate the association between gene expression in the prognostic model and the infiltration of various immune cells in breast cancer.Concurrently,the Clinical Letter Home (https://www.aclbi.com/static/index.html#/,accessed on July 24,2023) website was used to analyze the relationship between the gene expression and breast cancer prognosis models in the context of immune cell infiltration in different classifications.This analysis enables the exploration of the correlation between differential genes in the prognostic model and the tumor immune microenvironment,thereby providing insights into the potential interplay between the prognostic model and immune cell infiltration.
RESULTS
Construct the core mRNA characteristics of AICD
A comprehensive literature search was conducted using databases including PubMed(https://pubmed.ncbi.nlm.nih.gov/),Embase (Excerpta Medica Database,https://www.embase.com/),Web of Science (https://www.webofscience.com/),MESH (Medical Subject Headings,https://www.nlm.nih.gov/mesh/meshhome.html),and Scopus (https://www.scopus.com/),to identify a total of 35 mRNAs that are strongly associated with AICD (Table 1)[1,2,22,26,39-49].
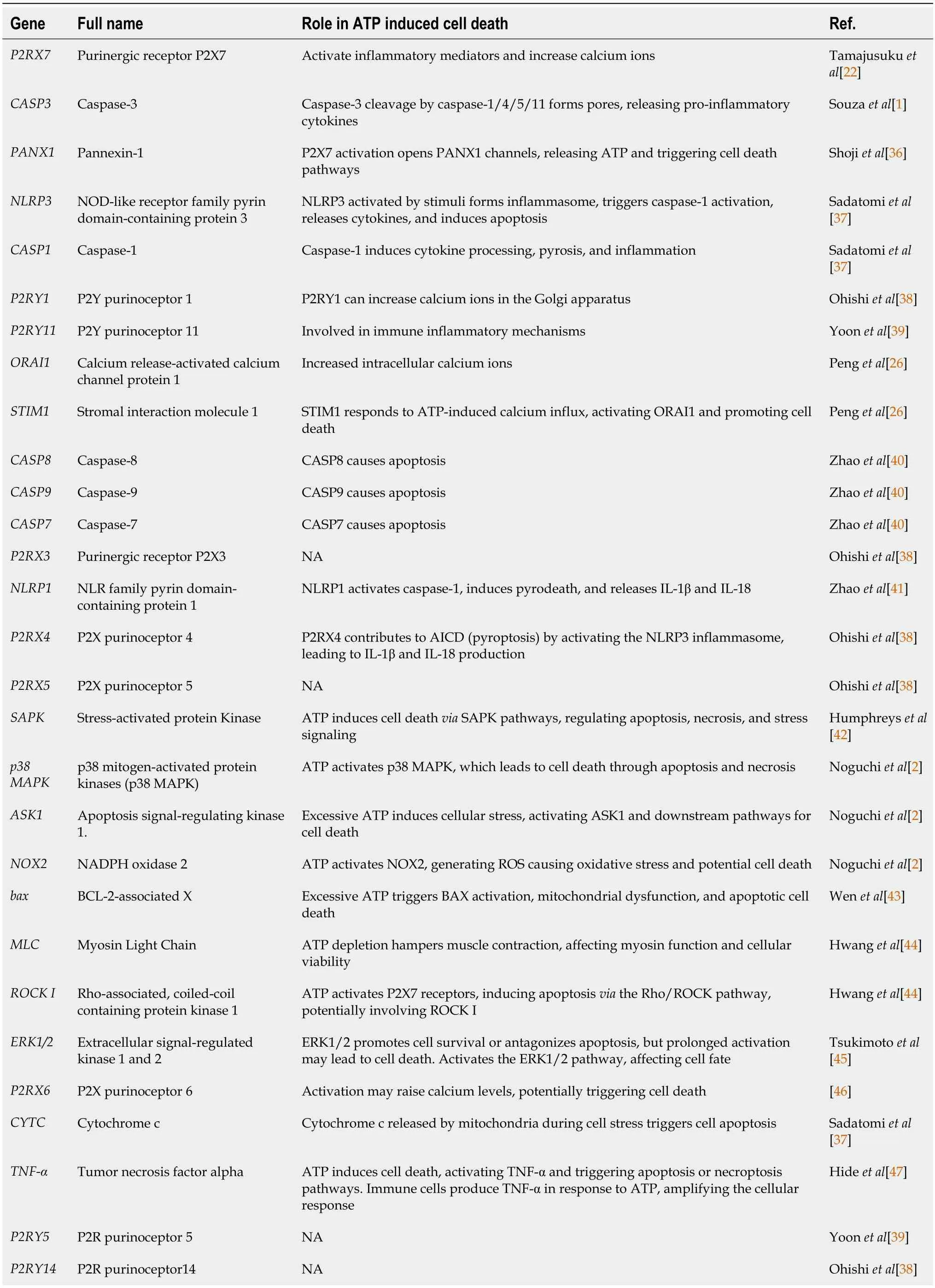
Table 1 List of core mRNAs of ATP-induced cell death

IL-18: Interleukin 18;IL-1b: Interleukin 1 beta;MAPK: Mitogen-activated protein kinase;NA: Not available.
According to the univariate Cox proportional hazard regression analysis,4 of the 35 mRNAs in the training cohort exhibited significantP< 0.05 (Figure 1A).Utilizing the LASSO-Cox regression analysis,the selection was further narrowed down to four mRNAs,which were incorporated into the prognostic model for the training cohort (Figure 1B and C).The construction of the new risk scoring formula based on these selected mRNA features was as follows: risk score=0.344 × pannexin 1 (PNAX1) -0.223 × P2RX4 -0.274 × caspase 7 (CASP7) -0.198 × cyclin D2 (CCND2).
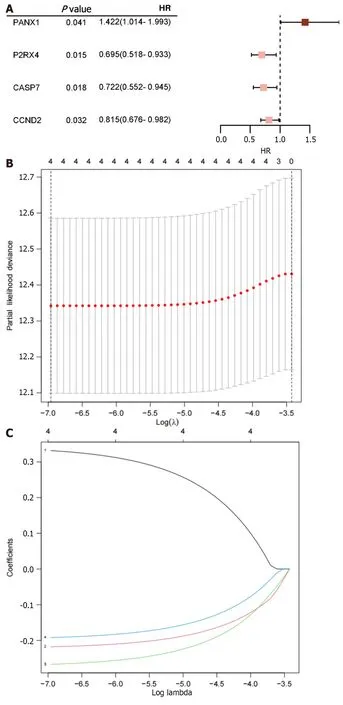
Figure 1 lllustrates the results obtained from the analyses. A: Four ATP-induced cell death-related mRNAs are presented with P < 0.05 along with their corresponding risk ratios derived from the univariate Cox proportional hazard regression analysis;B: The process of tuning parameter (λ) selection for overall survival-related mRNAs in the Least Absolute Shrinkage and Selection Operator (LASSO) model is shown;C: The four chosen mRNAs' LASSO coefficient spectra,and the vertical line shows the coefficient values that the LASSO algorithm chose.HR: Hazard ratio.
mRNA characteristics significantly distinguished between high- and low-risk groups
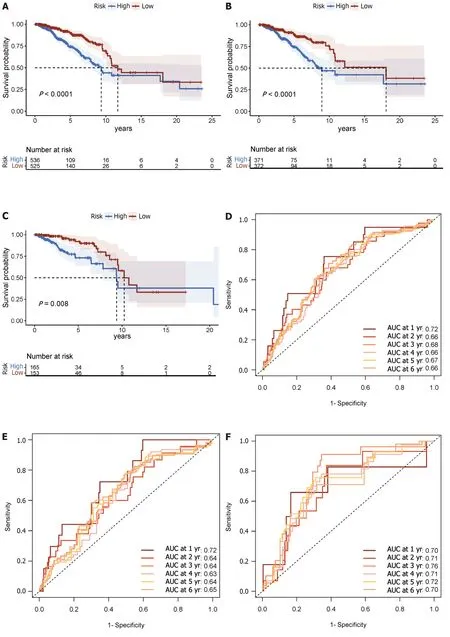
Each patient’s risk score was calculated using the accepted formula (Figure 2A).Subsequently,the robustness of these features was evaluated using training and internal validation cohorts (Figure 2B and C).Patients in the high-risk group had shorter OS than those in the low-risk group,according to KM survival analysis and bilateral log-rank tests performed on the complete dataset (P< 0.001;Figure 3A).

Figure 2 lllustrates the Cancer Genome Atlas Breast Cancer dataset entire dataset's risk score distribution and expression heat map. A1-A3: Total risk score;B1-B3: Training set risk score;C1-C3: Test set risk score;D1-D3: External validation cohort risk score;A1,B1,C1 and D1: The risk score distribution is illustrated,with pink dots representing the low-risk group and red dots representing the high-risk group.The vertical dotted line represents the median risk score cut-off point;A2,B2,C2 and D2: Displays the survival time and survival status of all patients;A3,B3,C3 and D3: Shows the expression patterns of the four selected genes from the ATP-induced cell death signature.

Figure 3 Displays time-varying receiver operating characteristic curves and Kaplan-Meier survival analyses. A: In the complete dataset,the Kaplan-Meier survival analysis curve revealed a substantial difference in overall survival (OS) among the low-risk and high-risk groups;B: As well as in the training;C: Validation cohorts;D: The time-varying receiver operating characteristic curve area under the curve values for the total dataset;E: Training group;F: Validation group at 5 yr of OS were 0.67,0.64,and 0.72,respectively;G: Kaplan-Meier survival analysis;H: Receiver operating characteristic curve over time.
The prognostic efficacy of this risk score was further validated in the test set and training set cohorts,wherein similar and significant OS differences were observed between the high-and low-risk groups (P< 0.05;Figure 3B and C).Additionally,for the 5-year OS prediction in the training and internal validation populations,the time-varying ROC curve analysis produced AUC values of 0.67,0.64,and 0.72,respectively (Figure 3D-F).
The prognostic significance of four mRNA features was verified in the METABRIC dataset
Further validation was conducted in the METABRIC dataset,comprising 1978 primary breast cancer cases.The established algorithm was used to compute the risk score for each patient.Subsequently,the robustness of the features was assessed in the validation queue of the METABRIC dataset (Figure 2D).
In the METABRIC dataset,significant differences were found in OS between the high-and low-risk groups,as had been seen in previous cohorts (P< 0.001;Figure 3G).Additionally,at 1,3,and 5 years,the time-varying ROC curve AUC values were 0.68,0.66,and 0.61,respectively (Figure 3H).
Construction of miRNA corresponding to the mRNA characteristics of the AICD core prognosis model
In this investigation,miRNA-mRNA target prediction was conducted utilizing the miRTarBase database as of July 21,2023.A comprehensive collection of 213 miRNAs was retrieved,after eliminating duplicate entries.Subsequently,through intersection analysis with the TCGA-BRCA and METABRIC databases,a refined set of 21 miRNAs was obtained(Table 2).

Table 2 MicroRNAs corresponding to the mRNA of a prognostic model
Construction of mRNA corresponding to the miRNA prognostic model of AICD
A total of 1019 primary breast cancer cases were extracted from the TCGA-BRCA datasetviathe UCSC Xena website,after excluding duplicate cases and those with incomplete OS data.Subsequently,these cases were randomly divided into a training set comprising 70% of the samples (n=714) and an internal validation set comprising 30% of the samples (n=305).
Six of the twenty-one miRNAs in the training group had univariate Cox proportional hazard regression analysis results withP< 0.15 (Figure 4A).Four of the miRNAs were subsequently chosen for inclusion in the training cohort's prognostic model using LASSO-Cox regression analysis (Figure 4B and C).The resulting risk scoring formula based on these features was as follows: risk score=0.181 × (hsa-miR-615-3p)+0.195 × (hsa-miR-324-3p) -0.333 × (hsa-miR-342-3p) -1.496 × (hsamiR-519b-3p).

Figure 4 Displays the results of the analysis on ATP-induced cell death-related microRNAs. A: Four microRNAs with P < 0.05 are provided,along with the risk ratios calculated using univariate Cox proportional hazard regression;B: Shows the selection process of tuning parameters (λ) for the overall survivalrelated miRNA using the Least Absolute Shrinkage and Selection Operator (LASSO) model;C: The four miRNAs' LASSO coefficient spectra are illustrated,with the vertical line reflecting the coefficient chosen using LASSO.HR: Hazard ratio.
miRNA profiles significantly distinguished between high- and low-risk groups
A risk score was calculated for each patient using the established formula (Figure 5A).To evaluate the dependability of these features,KM survival analysis and bilateral log-rank tests were performed on the training and internal validation cohorts (Figure 5B and C).The examination of the entire dataset cohort indicated an important distinction in OS among the high-risk and low-risk groups,with the high-risk group having a shorter OS (P< 0.001;Figure 6A).Furthermore,the predictive significance of this risk score was subsequently evaluated in both the test set and training set cohorts,revealing similarly substantial OS variations among the high-risk and low-risk groups (P< 0.05;Figure 6B and C).
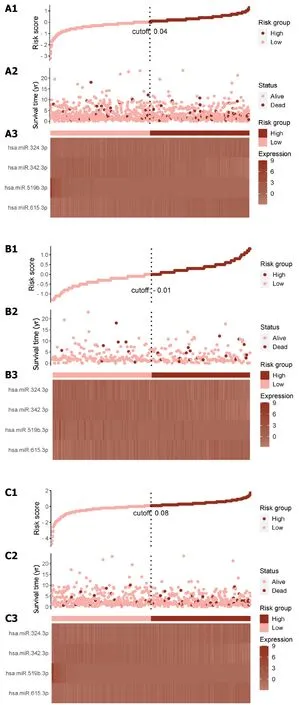
Figure 5 Risk score distribution and expression heat map for the entire Cancer Genome Atlas Breast Cancer dataset. A1-A3: Total risk score;B1-B3: Training set risk score;C1-C3: Test set risk score;D1-D3: External validation cohort risk score;A1,B1,C1 and D1: The risk score distribution is illustrated,with pink dots representing the low-risk group and red dots representing the high-risk group.The vertical dotted line represents the median risk score cut-off point;A2,B2,C2 and D2: Shows the patient's survival time and status;A3,B3,C3 and D3: Presents the heat maps depicting the expression levels of four selected ATPinduced cell death signature microRNAs.

Figure 6 Displays the Kaplan-Meier survival analysis as well as the receiver operating characteristic curve over time. A: In the total data set;B: Training cohort;C: Validation cohort,the Kaplan-Meier curve shows that the low-risk group had longer overall survival (OS) than the high-risk group;D: Timevarying receiver operating characteristic curve area under the curve for 5-yr OS was 0.70 for the total data set;E: 0.69 for the training group;F: 0.75 for the validation group.These findings illustrate the prognostic model's power in predicting patient outcomes.
The AUC for the time-varying ROC curve for the 5-year OS in the training and internal validation groups was 0.70,0.69,and 0.75,respectively (Figure 6D-F).
The prognostic significance of four miRNA features was verified in the METABRIC dataset
To further validate the prognostic model,validation was conducted using the METABRIC dataset,which comprised 1282 primary breast cancer cases.A risk score was calculated for each subject using the standard formula.Then the reliability and performance of the model were assessed in the METABRIC dataset validation cohort (Figure 5D).Consistent with prior findings,there were substantial differences in OS among high-and low-risk groups (P< 0.001;Figure 7A).For 1,3,and 5 years of OS,the time-varying ROC curve AUC values were 0.59,0.57,and 0.521,respectively (Figure 7B).These results affirm the robustness and prognostic value of the model in predicting patient outcomes in an independent dataset.

Figure 7 Displays time-varying receiver operating characteristic curves and Kaplan-Meier survival analyses. A: Kaplan-Meier survival analysis;B: Receiver operating characteristic curve over time.
Immune infiltration analysis of prognostic model genes
The validation of risk scores derived from the tissue-resident macrophage characteristics was confirmed by their strong correlation with the abundance of tumor-associated macrophage (TAM) subtypes within the dataset.Utilizing the CIBERSORT algorithm to estimate the immune-infiltrating population,it was observed that macrophages exhibited a positive correlation with all four selected mRNAs.Conversely,purity cells displayed the highest negative correlation with CASP7,CCND2,and PNAX1,with consistent correlations observed between these genes and immune infiltration(Figure 8).
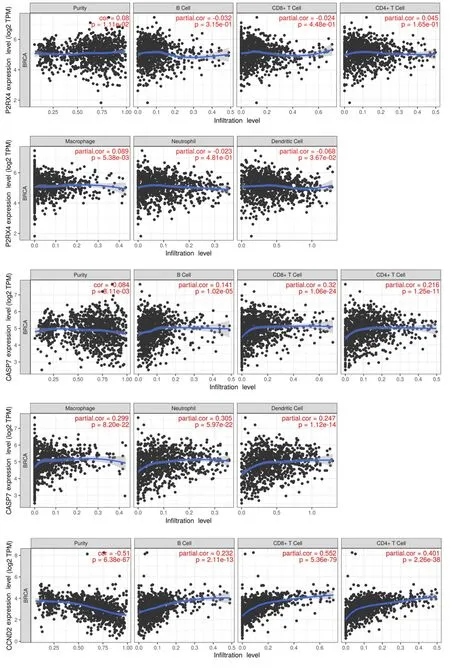
Figure 8 Correlation between mRNA and immune-infiltrated population.
mRNA differentiation between high- and low-risk differential gene expression and gene set enrichment analysis
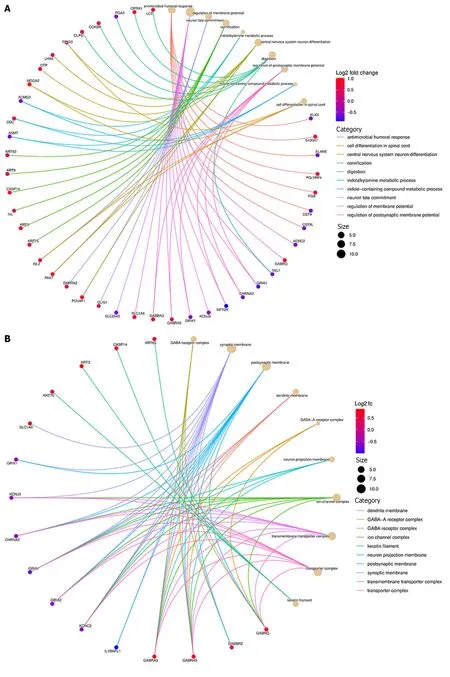
As indicated in Figure 9A below,169 genes were upregulated and 200 genes were downregulated in the high-low-risk group compared to the low-risk group (adjustedP=0.05 and |log2(fold change)| > 0.5).GO was used to conduct a representative analysis of differentially expressed genes,and the biological process (BP) enrichment results revealed that they were primarily involved in the control of systemic arterial blood pressure,amine transport,and neuropeptide signaling pathways (Figure 10).The results of cellular component (CC) enrichment showed that it was involved in terminal bouton,gamma-aminobutyric acid (GABA) receptor complex,and ion channel complex.The results of molecular function (MF) enrichment showed that it was involved in neuropeptide hormone activity and neuropeptide receptor binding.The KEGG pathway gene set enrichment analysis revealed that it was mostly engaged in neuroactive ligand receptor interaction,retinol metabolism,drug metabolism cytochrome P450,and xenobiotic metabolismviacytochrome P450.
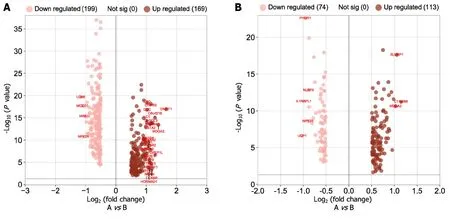
Figure 9 Volcano map of differential gene expression. A: Analysis of high-low risk difference in gene prognosis model;B: Analysis of high-low risk difference in the microRNA prognosis model.

Figure 10 Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analysis. A and E: Biological process (BP) enrichment results;B and F: Cell component (CC) enrichment results;C and G: Molecular function (MF) enrichment results;D and H: Kyoto Encyclopedia of Genes and Genomes enrichment results.
miRNA differentiating high-low risk differential gene expression and gene set enrichment analysis
In contrast with the low-risk group,as shown in Figure 9B below,differential gene expression revealed that 113 genes were upregulated,and 74 genes were downregulated (adjustedP=0.05 and |log2(fold change)| > 0.5).GO was used to conduct a representative analysis of differentially expressed genes.The BP enrichment results showed that they were involved in antimicrobial humoral response,regulation of membrane potential,and neuron fate commitment.Meanwhile,CC enrichment results showed that it was mainly involved in GABA receptor complex,synaptic membrane,and postsynaptic membrane,while MF enrichment was shown to be the main participant in extracellular ligand-gated ion channel activity,neurotransmitter receptor activity,and transmitter-gated ion channel activity.Lastly,the KEGG pathway gene set enrichment study revealed that it was mostly involved in neuroactive ligand receptor interaction,nicotine addiction,and morphine addiction (Figure 11).

Figure 11 Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analysis. A and E: Biological process (BP) enrichment results;B and F: Cell component (CC) enrichment results;C and G: Molecular function (MF) enrichment results;D and H: Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment results.
Immune infiltration analysis of risk scores in different breast cancer subtypes
Within the immune-infiltrating population estimated by the CIBERSORT algorithm,B cells,CD4+T cells,CD8+T cells,endothelial cells,and macrophages exhibited a consistent negative correlation with the risk score across all breast cancer types (Figure 12).Furthermore,the human epidermal growth factor receptor 2 (HER2)-enriched subtype showed a negative correlation with risk scores.Conversely,a positive connection was found between the risk score and natural killer (NK) cell infiltration in the HER2-positive,progesterone receptor (PR)-positive,luminal A,and normal-like subtypes.These results suggest that the risk score derived from the prognostic model is associated with distinct immune cell populations,indicating potential immunological implications for breast cancer progression and prognosis.

Figure 12 lmmune infiltration analysis of risk scores in different breast cancer types.A: Human epidermal growth factor receptor 2-positive(HER2+);B: HER2-;C: Estrogen receptor-positive (ER+);D: ER-;E: progesterone receptor-positive (PR+);F: PR-;G: Luminal A;H: Luminal B;I: HER2-enriched;J:Basal-like;K: Normal-like.
DlSCUSSlON
The subsets of cells that exhibited higher sensitivity to ATP expressed a greater number of P2X7Rs compared to the subsets of cells that were less sensitive.Consequently,interfering with the P2X7R reduced AICD,showing that it performs a significant role in this process.The primary mechanism of AICD appears to be necrosis,as evidenced by cellular atrophy with membrane permeabilization.However,there was no evidence of apoptosis,as defined by phosphatidylserine externalization and CASP activity[22].These findings highlight the importance of P2X7 in AICD and cast light on its role in tumor growth.In many inflammatory conditions,elevated levels of extracellular ATP function as mediators to activate the P2X7R.However,a distinct study demonstrated that ATP treatment induced apoptosis and autophagy-mediated cell death in the HCT8 cell line,rather than necrosis[1].In healthy human intestinal epithelial tissue explants,extracellular ATP may be the cause of CASP3 activation and apoptosis,which would explain the underlying process.This further stresses that AICD is a multifaceted process influenced by cell type and the specific microenvironment.
In modern society,breast cancer has become a major public health concern.Under the impact of different carcinogenic causes,the pathophysiology of this illness involves the unchecked proliferation of mammary epithelial cells,which is linked to cell death processes.Several classical modes of cell death,such as autophagy,apoptosis,and necrosis,as well as iron and copper death,have been extensively studied.AICD exhibits a degree of specificity while also displaying interplay with other cell death pathways.This phenomenon may result in the release and disturbance of iron and copper ions,consequently activating core pathways of autophagy,apoptosis,and necrosis,thereby augmenting the activation and execution of other cell death pathways.In turn,activation of cell death pathways such autophagy,apoptosis,and necrosis may alter ATP release and metabolic processes,influencing the outcome of AICD.However,the precise processes behind the key role of AICD are still unknown.
Hence,this paper proposes a hypothesis positing that AICD serves as a fundamental logic of cell demise,contributing to a deeper comprehension of disease treatment and progression.To support this hypothesis,a comprehensive review of relevant literature was carried out,focusing on the basic mechanisms and core genes involved in AICD.Additionally,functional mechanisms are explored within the context of breast cancer.This research endeavor sheds light on the intricacies of AICD and its potential implications in breast cancer management.
There is limited literature on the association between AICD and breast cancer,but research on the pivotal signaling receptor P2X7 in this context is more extensive.P2X7 is a purinergic ligand-gated ion channel receptor that functions as a non-selective cationic channel that is activated by ATP.This receptor is essential for signaling,growth regulation,cytokine release,and tumor cell formation[9].Recent research has demonstrated that overexpression of the P2X7R in breast cancer controls the v-akt murine thymoma viral oncogene homolog (AKT) signaling pathway,the calcium (Ca2+)-activated potassium channel subfamily K member 3 potassium channel,and the epithelial-mesenchymal transition(EMT),as well as controlling extracellular vesicle release.Consequently,these pathways drive breast cancer cell invasion and migration,and their modulation is influenced by variables such as hypoxia and ATP.Furthermore,miRNAs have been found to bind to the 3' untranslated region of the P2X7R,influencing the occurrence and progression of breast cancer by either upregulating or downregulating the receptor’s expression.In response to these discoveries,novel P2X7R inhibitors such as emodin and unicell have demonstrated the ability to hinder P2X7R-mediated breast cancer invasion,thus holding promise for potential clinical applications[3].Additionally,P2X7 has been found to be overexpressed in tamoxifen-resistant breast cancer cells,and functional investigations have been undertaken using selective P2X7 antagonists to study its effects[50].
miRNAs are important regulators of posttranscriptional gene expression,and they play a significant part in cellular processes including differentiation,proliferation,and metastasis.Notably,miR-150 is upregulated in both breast cancer cell lines and tissues.Inhibiting miR-150 function resulted in cell death in several breast cancer cell lines,whereas ectopic expression resulted in increased cell proliferation[18].These findings highlight the potential of miRNAs and mRNAs as novel therapeutic targets for breast cancer.
However,it is noteworthy that the current study did not include P2X7 in the predictive model,suggesting that P2X7 may not be a significant predictor of patient outcomes in breast cancer.Interestingly,the predictive model incorporated the P2X4R instead,which reveals new avenues for the treatment of breast cancer.
The mRNA prognostic model utilized in this study encompassed four specific mRNAs,namely P2X4,PNAX1,CASP7,and CCND2.P2X4 is a P2 receptor located on the cell membrane,which becomes activated when there is an excess of extracellular ATP.This activation then activates other P2 receptors,such as P2X7,as well as the PNAX1 channel,resulting in the extrusion of intracellular ATP from the cellviathe PNAX1 channel.Consequently,this process promotes AICD.Additionally,the activation of P2 receptors on the cell membrane can also stimulate immune-inflammatory factors,CASP7 apoptosis factors,CCND2,and other factors that influence the apoptosis and autophagy processes in cells.This perspective reinforces the concept of AICD as the fundamental basis of cellular demise,thereby distinguishing it from other modes of cell death.Nevertheless,it is worth noting that existing studies have yet to establish a direct relationship between AICD and cancer,especially in the context of breast cancer prognostic models.
The P2X4R is a member of the P2X receptor family that generates a highly Ca2+permeable non-selective cation channel on the plasma membrane in response to exogenous ATP.However,the P2X4R is distinguished from other subtypes by its preference for late endosomal,lysosomal,and/or lysosomal related organelles.The inclusion of tyrosine and dileucine transport motifs in the C-terminal and N-terminal domains,respectively,aids in this particular localization.Recent studies have shown that the P2X4R regulates ion flux on lysosomal membranes,modulating lysosomal membrane fusion and desensitization of receptors exposed to external ATP[51].
Recent investigations have highlighted the upregulation of P2X4 purinergic receptors in breast cancer patient biopsies,with a predominant localization in endolysosomes.Moreover,these investigations have shed light on the involvement of the P2X4R in boosting tumor development and metastasisin vivoand enhancing breast cancer cell invasion in vitro.The P2X4R’s pro-malignant impact is linked to its control of lysosomal acidity,promotion of autophagy,and facilitation of cell survival.Additionally,a correlation was observed between autophagy activity and EMT,with the influence of P2X4 being particularly pronounced under conditions of metabolic challenges.P2X4 gene silencing and pharmacological intervention inhibited autophagy and EMT,whereas restoring P2X4 expression in knockout cells restored the aggressive phenotype[52].These recent discoveries contribute valuable insights to our understanding of the underlying mechanisms involving the P2X4R in breast cancer and its regulatory role in autophagy and EMT processes.
The PANX protein channel family represents a class of channels characterized by a large pore size,serving as crucial mediators of intercellular communication with fundamental implications for cell development and maintenance of cellular homeostasis.Among these,PANX1 has been extensively studied and participates in regulating the permeability of ATP and ions within and outside cells,playing a pivotal role in various physiological processes and pathological mechanisms.Mutations in the PANX1 gene can impact protein glycosylation and accelerate ATP release,leading to cell death.
In breast cancer cells,the PANX1 channel exhibits a dual role in purinergic signaling,contributing to both cell survival and the induction of cytotoxic effects.This function is mediated by the amplified opening of the P2X4/P2X7-gated PANX1 channel,triggering cell death characterized by CASP1 activation and exhibiting a mixed pattern of apoptosis and necrosis,consistent with pyroptosis.Studies have revealed that cancer cell death is reliant on ATP release and downstream death signaling mediated by the P2X7R,which can be reversed by inhibiting NADPH oxidase-produced reactive oxygen species,Ca2+/calmodulin-dependent protein kinase II,or the mitochondrial permeability transition pore.Enhanced P2X4/P2X7 signaling may also be related with the ATP-rich tumor microenvironment,providing a molecular basis for purinergic receptor-mediated selective control of breast cancer and serving as a possible platform for comprehensive breast cancer immunotherapy[7].
CASP7,a member of the CASP family,plays a crucial role in the execution stage of apoptosis,wherein sequential CASP activation is vital.CASP exists as an inactive proenzyme,and proteolytic cleavage of conserved aspartic acid residues generates two large and small subunits,which then dimerize to form active enzymes.The precursors encoding for CASP7 protein are cleaved by CASP3 and CASP10,and they become activated upon stimulation by cell death signals,subsequently inducing apoptosis.
Recent research has investigated the expression of CASP7 in breast cancer patients,confirming its involvement with regulating tumorigenicity in breast cancer cells.Moreover,CASP7 expression was discovered to be higher in breast cancer tissues than in normal tissues.Ectopic expression of CASP7 has a significant connection with estrogen receptor alpha expression and demonstrated a progressive rise across various stages of breast tumor grading.According to KM analysis,increased levels of CASP7 expression were related with a better outcome in breast cancer patients following systemic endocrine therapy[53].Furthermore,according to the findings,both the precursor and active forms of CASP7 are mostly found outside of the cytoplasmic region of breast cancer cells,primarily within the nucleus.
Compared to comparable normal tissues and samples from healthy people,hypermethylation of the CCND2 promoter was seen in 40.9% of breast tumor patients and 44.4% of circulating cell-free DNA in plasma.Furthermore,the study showed that in female breast cancer patients,CCND2 promoter hypermethylation acts as a distinctly poor prognostic factor.Experiments using cell models clarified the functional involvement of CCND2 in breast cancer.Specifically,cancer cell proliferation and migration are reportedly inhibited by the expression of CCND2.Moreover,the demethylating drug antroquinonol D causes an increase in CCND2 expression,leading to cell cycle arrest and suppression of cancer cell growth and migration[54].
Furthermore,to enhance the prognostic model's precision to accurately predict patients' survival times and identify potential therapeutic targets,this study extended its investigation to model the miRNAs that correspond to the predictive genes identified after establishing the mRNA prognostic model.This iterative approach enabled the narrowing down of potential targets for future anticancer interventions,consequently augmenting the model's predictive accuracy.The miRNA prognostic model integrated four key miRNAs,namely hsa-miR-615-3p,hsa-miR-519b-3p,hsa-miR-342-3p,and hsa-miR-324-3p.These miRNAs were found to exert considerable influence on the prognosis of breast cancer,offering promising avenues for novel therapeutic targets and approaches in future breast cancer treatments.
Recent investigations have demonstrated the significant upregulation of miR-615-3p expression in both breast cancer cells and tissues,particularly in those with metastatic properties.Notably,stable overexpression of miR-615-3p in breast cancer cell lines resulted in notable enhancements in cellular motilityin vitro,along with a consequential increase in lung metastasisin vivo.The expression of mesenchymal markers increased along with these effects,and epithelial markers decreased.Further investigation found that reintroducing miR-615-3p resulted in increased downstream signaling of the transforming growth factor pathwayviatargeting the 3'-untranslated region (3'-UTR) of protein interacting with C Kinase 1 (PICK1).PICK1 inhibits the interaction of dicer 1,ribonuclease III and mothers against decapentaplegic homolog 2/3,as well as the processing of pre-miR-615-3p to its mature miRNA form,within breast cancer cells.This regulatory mechanism establishes a negative feedback loop[55].
Furthermore,the investigation revealed the noteworthy upregulation of HOXA transcript at the distal tip (HOTTIP)expression and the significant downregulation of miR-615-3p expression in breast cancer patients compared to healthy individuals,as well as patients from non-breast cancer groups (P< 0.001 for all comparisons).Importantly,in the serum of breast cancer patients,there was a substantial negative connection between the expression levels of HOTTIP and miR-615-3p[56].Additionally,by inhibiting CUGBP Elav-like family member 2 expression,miR-615-3p was discovered to increase cancer cell motility and proliferation[57].
Although research on the effect of miR-519b-3p on breast cancer is limited,it appears to have a major impact on cancer cell proliferation and invasion.Previous research has found that miR-519b-3p expression is significantly downregulated in colorectal cancer tissues and cell lines.Overexpression of miR-519b-3p inhibits the proliferation and invasion of colorectal cancer cell lines,the Rous sarcoma oncogene-transformed rat kidney cell line,and Duke's type B colorectal adenocarcinoma cell line 1.On the other hand,low expression has the reverse effect.
Notably,miR-519b-3p suppresses colorectal cancer cell proliferation and invasion by targeting the uMtCK/Wnt signaling pathway[58].Furthermore,LINC01419 negatively regulates miR-519b-3p,with renal cell carcinoma differentiation 1 (RCCD1) identified as a downstream target of miR-519b-3p and its expression being negatively associated with that of miR-519b-3p.Conversely,the expression of LINC01419 is positively correlated with RCCD1 expression.Later studies showed that the effects of LINC01419 silencing on cell proliferation,migration,and invasion could be mitigated by overexpressing miR-519b-3p inhibitors or RCCD1.These findings suggest that LINC01419 enhances RCCD1 expression through sequestering miR-519b-3p,thereby promoting carcinogenesis in lung adenocarcinoma.As a result,LINC01419 holds potential therapeutic value in the management of lung adenocarcinoma[59].
MiR-342-3p has been linked to a poor prognosis and decreased expression in triple-negative cancers.Experimental findings have shown that the proliferation,viability,and motility of several triple-negative tumor cellsin vitroare greatly decreased when miR-342-3p is overexpressed.MiR-342-3p regulates lactic acid and glucose fluxes by directly targeting monocarboxylate transporter 1,causing the metabolic equilibrium of tumor cells to be disrupted.Additionally,cells with elevated miR-342-3p levels demonstrate higher optical reduction-oxidation rates[60].The expression of miR-342-3p in breast cancer cells is significantly reduced in patients with metastatic disease.Inhibiting miR-342-3p inhibits aggressive and drug-resistant activity in breast cancer tumor cells.Further investigations have confirmed the presence of binding sites between miR-342-3p and inhibitor of DNA binding 4 (ID4).Notably,ID4 reduces the effects of miR-342-3p on chemotherapy resistance.ID4 small interfering RNA has also been shownin vivoto have tumor-suppressive properties.MiR-342-3p suppresses metastasis and chemotherapy resistance in breast cancer cells by targeting ID4,indicating its potential as a tumor suppressor[61].This discovery identified miR-342-3p as a possible therapeutic target for breast cancer treatment.
Metformin causes iron mortality in MDA-MB-231 cellsviaincreasing the expression of miR-324-3p.Overexpression of miR-324-3p leads to the inhibition of cancer cell growth activity,while its inhibition promotes cell growth activity.The regulatory action of miR-324-3p involves direct targeting of glutathione peroxidase 4 (GPX4)viabinding to the 3'-UTR region of GPX4,resulting in the downregulation of GPX4 expression[62].MiR-324-3p may exert its regulatory role through multiple pathways,including iron death and AICD.LINC00963 works by blocking the inhibitory activity of miR-324-3p on activated Cdc42-associated kinase 1 (ACK1) expression.Clinically,miR-324-3p has a negative correlation with LINC00963 expression in breast cancer tissues.The inhibitory effect of miR-324-3p on the growth and radiosensitivity of breast cancer cells can be reduced by overexpressing LINC00963 or ACK1[63].
Additionally,in the tissues and cells of triple-negative breast cancer,small nucleolar RNA host gene 22 (SNHG22) is abundantly expressed.Triple-negative breast cancer cells are prevented from proliferating,migrating,and invading when SNHG22 is silenced.Furthermore,SNHG22 acts as a sponge for miR-324-3p,lowering its expression in triple-negative breast cancer cells.However,miR-324-3p upregulation suppresses the proliferation and migration of triple-negative breast cancer cells[64].Notably,miR-324-3p inhibition,or suppressor of defective silencing 3 overexpression can reverse the anticancer effect of SNHG22 silencing on the malignant phenotype of triple-negative breast cancer cells.
The CIBERSORT algorithm was used to quantify immune infiltration,and there was a substantial association between immune-infiltrating populations and risk scores in distinct breast cancer subtypes.B cells,CD4+T cells,CD8+T cells,endothelial cells,and macrophages exhibited an inverse association with risk scores across all breast cancer subtypes.Moreover,the HER2-enriched subtype showed a negative correlation with risk scores.These findings suggest a potential protective effect or involvement of these immune cell populations in the immune response,leading to a reduced risk and improved prognosis in breast cancer patients.Conversely,in the HER2-positive,PR-positive,luminal A,and normal-like classifications,there was a positive correlation with the infiltration of NK cells.This shows that NK cells may have a role in or contribute to the disease genesis and progression,significantly impacting patient outcomes.Furthermore,macrophages showed a positive correlation with the expression of P2X4,PNAX1,CASP7,and CCND2.This association may indicate a potential link between macrophage infiltration and the activation of specific genes associated with breast cancer.
NK cells are critical components of the anti-tumor surveillance system,and in animal models,they serve a significant part in suppressing tumor growth and metastasis.The reduction in natural killing activity has been positively associated with the extent of tumor dissemination[65].This bears substantial relevance in the context of tumors,as transformation events may lead to the downregulation of auto ligands and stress-induced ligands that are recognized by NK cells.Additionally,NK cell activation results in the secretion of stimulatory cytokines,which participate in cancer elimination through various direct mechanisms and by stimulating the adaptive immune system[65].The Janus kinase (JAK)/signal transducer and activator of transcription pathway remains active during metastasis;therefore,the impact of JAK inhibitors on NK cells and the potential value of immunostimulants to enhance weakened tumor immune surveillance merit consideration in the clinical management of breast cancer[66].
Macrophages exhibit distinct activation states and functions,and can be categorized into M1 type (classically activated macrophages) and M2 type (alternatively activated macrophages).TAMs bear similarities to M2 macrophages.Clinicopathological studies have demonstrated that TAMs are associated with a poor clinical prognosis in tumors.In the context of human breast cancer,a high density of TAMs is linked to an unfavorable prognosis.Extensive research has revealed the role of TAMs in breast cancer progression.This encompasses the capacity of TAMs to induce angiogenesis,promote invasion through remodeling the tumor extracellular matrix,mimic breast cancer cells to evade the host immune system,and recruit immunosuppressive white blood cells to the tumor microenvironment.In breast cancer,TAMs can also enhance cancer cell invasion and suppress the anti-tumor function of tumor-suppressing T cells.This leads to a compromised anti-tumor immune response and subsequently impacts patient prognosis[67].
KEGG analysis showed that the genes differentiated by mRNA risk score significantly enriched 25 signaling pathways.Also,those genes differentiated by miRNA risk score significantly enriched 29 signaling pathways,and the two genes jointly enriched 16 signaling pathways.These include neuroactive ligand-receptor interaction,retinol metabolism,drug metabolism-c cytochrome P450,pancreatic secretion,metabolism of xenobiotics by cytochrome P450,nicotine addiction,porphyrin metabolism,chemical carcinogenesis-DNA adducts,the cAMP signaling pathway,ascorbate and aldarate metabolism,GABAergic synapse,steroid hormone biosynthesis,pentose and glucuronate interconversions,protein digestion and absorption,nitrogen metabolism and drug metabolism -other enzymes.The enrichment analysis results show that the model has satisfactory performance.

The neuroactive ligand-receptor interaction signaling pathway represents the highest enrichment in both models.This pathway encompasses the intricate interactions between neuroactive substances within the nervous system,such as dopamine,acetylcholine,glutamate,serine,GABA,and their corresponding receptors.These interactions elicit a range of regulatory mechanisms,including protein kinase cascades,G-protein-coupled receptor signaling,and modulation of intracellular calcium concentration.
Based on the findings,increased usage of dopamine antagonists for the treatment of mental disorders may be linked to the development and promotion of breast cancer.The use of antipsychotic dopamine antagonists demonstrated a doseresponse relationship with cumulative dose,resulting in a 16% increased risk of breast cancer[68].Extracellular ATP has recently emerged as a crucial factor in regulating pathological cell death.Exposure to ATP induces cell death characterized by cell swelling,disruption of endoplasmic reticulum integrity,formation of large cytoplasmic vacuoles,and ultimately cell lysis and DNA release.
Additionally,ATP processing triggers the cleavage of CASP3,a hallmark of apoptosis.This suggests that the release of extracellular ATP from damaged tissues may induce dopaminergic cell death by accelerating necrosis of P2X7Rs[69].PNAX1 participates in the release of ATP and glutamate in neurons and astrocytes.Extracellular ATP activates PNAX1 at the resting membrane potential through the P2X7R.Studies have indicated that the P2X7R-PNAX1 complex may function as a negative regulator mediated by acetylcholine receptorsin vivo[70].A recent study demonstrated the promising potential of using low-concentration muscarinic receptor agonists in combination with conventional chemotherapy agents for breast cancer treatment[71].In summary,extracellular ATP plays a role in the neuroactive ligand-receptor interaction signaling pathway,influencing the invasion,survival,and migration of breast cancer cells.
In general,the interpretation of the data in this study may be subject to certain limitations,warranting the need for prospective experimental validation to enhance the reliability of the research findings.Initially,an extensive literature search was conducted to identify the key mechanisms of AICD that have been experimentally verified.Building upon this foundation,the correlation between AICD and breast cancer prognosis has been elucidated for the first time.Consequently,a prognostic model has been developed that integrates mRNA and miRNA,effectively narrowing down potential therapeutic targets for future anticancer interventions and augmenting the precision of the predictive model.
CONCLUSlON
This study conducted a comprehensive investigation into the primary mechanism underlying AICD and performed an indepth analysis of the associated mRNA expression patterns.Notably,mRNA and miRNA characteristic models were successfully established and specifically tailored to AICD,both of which exhibit potential as independent prognostic factors.Leveraging these two models achieved heightened precision in estimating patient survival status and simplified the decision-making process regarding relevant therapeutic interventions.Consequently,the findings offer a robust scientific foundation for comprehending the fundamental logic governing cell death.Moreover,the clinical implications of this research are highly significant,as they shed light on the regulatory mechanisms of cell death and provide valuable guidance for the treatment and prognosis evaluation of breast cancer.Future investigations need to allocate greater focus on the examination,analysis,and discourse of discrete cancer cells,in order to reach more exacting insights.Recognizing the fact that the domain of cancer research is inherently rooted in single-cell substrates,it is imperative to note that a dearth of single-cell analyses for individual patients could potentially undermine the comprehensive nature of these studies.Subsequent to this,the primary objectives include giving precedence to functional insights at the single-cell level,thereby ensuring that later research provides actionable and targeted insights.
ARTlCLE HlGHLlGHTS
Research background
ATP-induced cell death (AICD) is a unique mode of cell death caused by high levels of extracellular ATP and is closely linked to various cancer advancements.It has a dual impact on breast cancer by participating in the regulation of death pathways and leading to increased extracellular ATP levels,creating an interconnected regulatory loop.AICD is a pivotal mechanism regulated by genes and microRNAs (miRNAs),contributing to breast cancer progression.However,the precise nature of their interactions requires further research.Manipulating ATP levels and receptors can alter breast cancer cell proliferation,invasion,and metastasis,highlighting the potential for AICD in breast cancer treatment.AICD is a multifaceted process,with overarching mechanisms including purinergic receptor activation,particularly P2X purinoceptor 7 receptor (P2X7R),elevation of intracellular calcium ion concentration,inflammatory responses,and mitochondrial function disruption.miRNAs are key regulators of gene expression,functioning as either tumor suppressors or promoters,and their expression can be influenced by ATP levels.The relationship between AICD and miRNA involves two distinct mechanisms: miRNA targeting of essential genes in ATP-related signaling pathways and alterations in ATP levels influencing miRNA expression.Prognostic models,both gene and miRNA-based,are valuable for personalized treatment and prognosis assessment despite their limitations.The concurrent use of both models may provide a more comprehensive understanding of cancer prognosis.
Research motivation
No studies to date have investigated the impact of AICD regulatory mechanisms in breast cancer.Therefore,the primary goal of this paper was to look into the potential prognostic importance of AICD genes in breast cancer,as well as the interaction among AICD genes and prognostic gene-associated miRNAs in breast cancer.
Research objectives
This study conducted a comprehensive investigation into the primary mechanism underlying AICD and performed an indepth analysis of the associated mRNA expression patterns.Notably,mRNA and miRNA characteristic models were successfully established and specifically tailored to AICD,both of which exhibit potential as independent prognostic factors.Leveraging these two models achieved heightened precision in estimating patient survival status and simplified the decision-making process regarding relevant therapeutic interventions.Consequently,the findings offer a robust scientific foundation for comprehending the fundamental logic governing cell death.Moreover,the clinical implications of this research are highly significant,as they shed light on the regulatory mechanisms of cell death and provide valuable guidance for the treatment and prognosis evaluation of breast cancer.
Research methods
The foundational genes orchestrating AICD mechanisms were extracted from the scholarly literature,underpinning the establishment of a prognostic model.Simultaneously,a miRNA prognostic model was constructed that mirrored the gene-based prognostic model.Distinctions between high and low-risk cohorts within mRNA and miRNA characteristic models were scrutinized,with the aim of delineating common influence mechanisms,substantiated through enrichment analysis and immune infiltration assessment.
Research results
The mRNA prognostic model in this study encompassed four specific mRNAs—P2X4,pannexin 1,caspase 7,and cyclin D2.The miRNA prognostic model integrated four pivotal miRNAs: hsa-miR-615-3p,hsa-miR-519b-3p,hsa-miR-342-3p,and hsa-miR-324-3p.B cells,CD4+T cells,CD8+T cells,endothelial cells,and macrophages exhibited inverse correlations with risk scores across all breast cancer subtypes.Furthermore,Kyoto Encyclopedia of Genes and Genomes analysis revealed that genes differentially expressed in response to mRNA risk scores significantly enriched 25 signaling pathways,while miRNA risk scores significantly enriched 29 signaling pathways,with 16 pathways being jointly enriched.
Research conclusions
This study conducted a comprehensive investigation into the primary mechanism underlying AICD and performed an indepth analysis of the associated mRNA expression patterns.Notably,mRNA and miRNA characteristic models were successfully established and specifically tailored to AICD,both of which exhibit potential as independent prognostic factors.Leveraging these two models achieved heightened precision in estimating patient survival status and simplified the decision-making process regarding relevant therapeutic interventions.Consequently,the findings offer a robust scientific foundation for comprehending the fundamental logic governing cell death.Moreover,the clinical implications of this research are highly significant,as they shed light on the regulatory mechanisms of cell death and provide valuable guidance for the treatment and prognosis evaluation of breast cancer.
Research perspectives
Future investigations need to allocate greater focus on the examination,analysis,and discourse of discrete cancer cells,in order to reach more exacting insights.Recognizing the fact that the domain of cancer research is inherently rooted in single-cell substrates,it is imperative to note that a dearth of single-cell analyses for individual patients could potentially undermine the comprehensive nature of these studies.Subsequent to this,the primary objectives include giving precedence to functional insights at the single-cell level,thereby ensuring that later research provides actionable and targeted insights.
FOOTNOTES
Co-corresponding authors:Sandai Doblin and Song Zhi-Jing.
Author contributions:Zhang HL was responsible for drafting the primary manuscript and conducting the data analyses;Dinesh B,Tabana Y,Saad DS,Adam Ahmed Adam M,Zhang HL,Zhao R,Barakat K,Wang Y,and Wang W were involved in the data collection,and preparation of tables and charts;Sandai D,Zhang ZW,and Song ZJ had pivotal roles in the research design,guiding the research group,and orchestrating the collaborative efforts of all authors;Sandai D and Song ZJ gave detailed guidance on the paper;All authors have read and approved the final manuscript.
Supported byNational Natural Science Foundation of China,No.81 960877;University Innovation Fund of Gansu Province,No.2021A-076;Gansu Province Science and Technology Plan (Innovation Base and Talent Plan),No.21JR7RA561;Natural Science Foundation of Gansu Province,No.21JR1RA267 and No.22JR5RA582;Education Technology Innovation Project of Gansu Province,No.2022A-067;Innovation Fund of Higher Education of Gansu Province,No.2023A-088;Gansu Province Science and Technology Plan International Cooperation Field Project,No.23YFWA0005;and Open Project of Key Laboratory of Dunhuang Medicine and Transformation of Ministry of Education,No.DHYX21-07,No.DHYX22-05,and No.DHYX21-01.
lnstitutional review board statement:No experimental studies on human and animal ethics were designed in this study.
lnformed consent statement:This study did not conduct clinical trials and did not involve informed consent signed by subjects and investigators.
Conflict-of-interest statement:The authors have no conflicts of interest to declare.
Data sharing statement:Technical appendix,statistical code,and dataset available from the corresponding author at [doblin@usm.my].Participants gave informed consent for data sharing.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the
original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Malaysia
ORClD number:Sandai Doblin 0000-0003-0544-4260;Zhi-Jing Song 0009-0002-3991-2907.
S-Editor:Lin C
L-Editor:Filipodia
P-Editor:Zhang XD
 World Journal of Clinical Oncology2024年2期
World Journal of Clinical Oncology2024年2期
- World Journal of Clinical Oncology的其它文章
- Unlocking the potential-vitamin D in prostate cancer prevention
- Updates on management of gliomas in the molecular age
- Deregulation of interferon-gamma receptor 1 expression and its implications for lung adenocarcinoma progression
- ldentification of immune cell-related prognostic genes characterized by a distinct microenvironment in hepatocellular carcinoma
- Population-based X-ray gastric cancer screening in Hiroshima prefecture,Japan
- Endoscopic resection for calcifying fibrous tumors of the gastrointestinal tract