
新生儿筛查C5-OH异常确诊2例3-甲基巴豆酰辅酶A羧化酶缺乏症*
2024-03-06 07:55曾君姜盼盼张润玲王小丽吴莉萍纪国富吕杰杨江涛吴本清中国科学院大学深圳医院光明医学实验中心广东深圳806深圳罕见病代谢组学精准医学工程研究中心广东深圳807深圳爱湾医学检验实验室广东深圳807长春市妇幼保健院计划生育服务中心长春0000葫芦岛市妇幼保健院计划生育服务中心辽宁葫芦岛000
临床检验杂志 2024年1期
曾君,姜盼盼,张润玲,王小丽,吴莉萍,纪国富,吕杰,杨江涛,吴本清(.中国科学院大学深圳医院(光明)医学实验中心,广东深圳 806;.深圳罕见病代谢组学精准医学工程研究中心,广东深圳 807;.深圳爱湾医学检验实验室,广东深圳 807;.长春市妇幼保健院计划生育服务中心,长春 0000;.葫芦岛市妇幼保健院计划生育服务中心,辽宁葫芦岛 000)
临床上3-羟基异戊酰肉碱(C5-OH)升高可提示多种遗传代谢病[1],其中,3-甲基巴豆酰辅酶A羧化酶缺乏症(3-methylcrotonylcoenzyme A carboxylase deficiency, MCCD)是最常见的血液C5-OH升高的遗传代谢病之一[2]。欧美地区报道,在新生儿串联质谱筛查中MCCD的发病率约为1/36 000[3],国内报道的发病率为1/68 333[4]。MCCD的临床表现变化较大,多数患者无临床表征,也有患者表现出严重的神经系统受损,甚至死亡。根据临床表现主要分为三类:无症状、母源性及有症状。有症状者可以通过限制亮氨酸或蛋白质等诱因饮食,予以高糖保证热量及各种营养素供应,如出现严重肉碱缺乏,可以进行L-肉碱补充,而对应无症状者不需要治疗[1]。
MCCD(OMIM 210200/210210)是一种常染色体隐性遗传病(AR),致病基因主要是位于第3号染色体长臂(3q25-q27)上的MCCC1(methylcrotonyl-CoA carboxylase subunit 1)基因和第5号染色体长臂(5q12-q13)上的MCCC2(methylcrotonyl-CoA carboxylase subunit 2)基因[5]。其发病机制是由于3-甲基巴豆酰辅酶A羧化酶活性异常致使3-甲基巴豆酰辅酶A堆积,3-甲基巴豆酰辅酶A与甘氨酸结合生成3-甲基巴豆酰甘氨酸(3-methylcrotonylglycine,3-MCG),使尿液中3-MCG和3-羟基异戊酸(3-hydroxyisovaleric,3-HIVA)浓度升高[1]。本研究旨在探讨新生儿筛查C5-OH异常联合尿有机酸、基因突变检测对疾病的鉴别诊断价值。报道如下。
1 材料与方法
1.1研究对象 选取2019年10月至2022年9月在中国科学院大学深圳医院(光明)进行遗传代谢病筛查的14例滤纸干血斑初筛C5-OH浓度高于阳性临界值(0.45 μmol/L)的新生儿为研究对象,其中男2例,女12例,但仅7例新生儿家长同意进行基因检测。本研究通过中国科学院大学深圳医院(光明)医学伦理委员会审核批准(批准文号:LL-KT-2021163),患儿家属知情同意。
1.2主要仪器及试剂 衍生化多种有机酸检测试剂盒(深圳爱湾智造科技有限公司),肌酐检测试剂盒(深圳普门科技股份有限公司),新生儿遗传代谢病筛查试剂盒(广州丰华生物股份有限公司),甲醇、乙酸乙酯(4 L/瓶,色谱纯)购自上海安谱公司、MultipSeq®Custom Panel文库构建试剂盒[艾吉泰康(嘉兴)生物科技有限公司]。GCMS QP 2020 NX型气相质谱联用仪(日本岛津公司),H1650R台式高速离心机、尿微量蛋白肌酐生化仪、恒温培养箱(东莞谱标科技公司),氮气吹扫仪 (杭州瑞诚仪器公司),Qubit®3.0荧光定量仪(美国Thermo Fisher Scientific公司),Aglient 2100生物分析仪(美国Agilent公司), MGI 2000高通量测序平台(深圳华大智造科技股份有限公司),串联质谱仪(API 3200MD,美国AB SCIEX公司)。
1.3方法
1.3.1样本收集 使用枸橼酸钠真空抗凝管采集新生儿非空腹静脉血1.0 mL, 4 ℃保存备用。干燥洁净容器采集新生儿新鲜随机尿液,分装至容量为10 mL的带盖无菌刻度离心管中,置于-20 ℃保存备用。
1.3.2串联质谱法(MS/MS)检测C5-OH浓度 按照串联质谱仪(API 3200MD)及新生儿遗传代谢病筛查试剂盒说明书操作检测样本中氨基酸和酰基肉碱含量。其中,C4DC+C5-OH参考区间:0.06~0.45 μmol/L,(C4DC+C5-OH)/C0参考区间:0.01~0.03,(C4DC+C5-OH)/C8参考区间:0.50~12.85。
1.3.3气相色谱-质谱联用技术(GC-MS)检测尿液有机酸水平 按照衍生化多种有机酸检测试剂盒说明书将待测尿液经除尿素、添加内标、肟化、液液萃取、氮吹、衍生等处理后,使用气相色谱-质谱联用仪测定132种尿液有机酸水平,通过测定有机酸目标物的峰面积与内标物十七烷酸峰面积求得比值×100,得到半定量结果[6]。其中,针对3-甲基巴豆酰辅酶A羧化酶缺乏症可参考3-羟基异戊酸-2(参考区间:0.00~6.10)、3-甲基巴豆酰甘氨酸-1(参考区间:0.00~1.05)、3-甲基巴豆酰甘氨酸-2(参考区间:0.00~0.52)含量;生物素酶缺乏症和全羧化酶合成酶缺乏症可参考3-羟基丙酸-2(参考区间:0.00~1.95)、甲基枸橼酸-4(参考区间:0.00~1.81)、3-甲基巴豆酰甘氨酸-1、3-甲基巴豆酰甘氨酸-2含量;3-羟基-3-甲基戊二酸尿症可参考3-羟基-3-甲基戊二酸-3(参考区间:0.00~30.52);3-甲基戊烯二酸尿症可参考3-甲基戊烯二酸-2(参考区间:0.00~0.50)、3-甲基戊二酸-2(参考区间:0.00~10.48)含量;β-酮硫解酶缺乏症可参考2-甲基-3-羟基丁酸-2(参考区间:0.00~7.65)含量。
1.3.4高通量测序技术检测基因突变 提取血液样本DNA,按照MultipSeq®Custom Panel文库构建试剂盒进行第一轮多重PCR扩增(T1和T2运行 95 ℃ 3 min 30 s;98 ℃ 20 s,60 ℃ 10 min,22个循环s;72 ℃ 5 min),纯化产物后[纯化磁珠(IGTTMPure Beads)+磁力架]再次进行PCR连接测序接头(95 ℃ 3 min 30 s;98 ℃ 20 s,58 ℃ 1 min,72 ℃ 30 s,9个循环;72 ℃ 5 min),纯化获得目标区域序列文库[纯化磁珠(IGTTMPure Beads)+磁力架],使用Qubit®3.0荧光定量仪与Aglient 2100生物分析仪进行文库质控;质控通过后利用MGI 2000高通量测序平台完成测序数据收集,获得平均测序深度≥100×,测序深度大于20×且达到95%以上的为有效数据。
1.3.5生物信息学寻找突变位点 数据通过BWA(Burrows-Wheeler Aligner)[7]软件比对到人参考基因组(Human_B37),使用GATK(Genome Analysis ToolKit)[8]分析变异信息、ANNOVAR软件进行变异注释[9],包括dbSNP数据库、千人基因组计划和其他已有的数据库的注释信息,注释内容涵盖人群频率、变异类型、功能预测(SIFT、Polyphen2、Mutation Taster等)、HGMD/Clinvar等致病性分类信息。变异致病性解读基于《遗传变异分类标准与指南(2017)》[10],共分为“致病”、“可能致病”、“意义未明”、“可能良性”及“良性”五类。
2 结果
2.1血液C5-OH和尿有机酸检测结果 14 例新生儿血液 C5-OH复检浓度均高于阳性临界值(0.45 μmol/L),滤纸干血斑初筛C5-OH的浓度区间为0.60~9.24 μmol/L,复检血液C5-OH的浓度区间为0.57~11.61 μmol/L。超过50%(8/14)的复检病例血液C5-OH浓度出现回落。除病例11与病例4之外,其余样本尿液有机酸检测3-甲基巴豆酰甘氨酸(3-MCG)与3-羟基异戊酸(3-HIVA)浓度正常;病例11初检和复检C5-OH浓度均超出阳性临界值20倍以上,尿液有机酸中3-羟基异戊酸(171.45 μmol/L),3-甲基巴豆酰甘氨酸(43.08 μmol/L)升高,结合血液中氨基酸及酰基肉碱的分析结果,提示3-甲基巴豆酰辅酶A羧化酶缺乏症。见表1。

表1 血液酰基肉碱和尿液有机酸检测结果
2.2基因突变检测结果 参与基因突变检测的7例新生儿在6个基因上共检测出2种突变类型和12个突变位点(表2)。其中在MCCC1与MCCC2基因检测出6个突变位点,均为杂合突变,分别为c.1518delG,c.-102C>T,c.1331G>A,c.1732-1G>C,c.470A>G,c.1103G>A,其中c.1518delG突变等级为致病,c.1331G>A和c.1732-1G>C为疑似致病,其余位点为意义未明,MCCC1基因位点(3:182817330*,NM_020166.3,c.-102C>T)为新发突变,可以补充突变与疾病的关系。

表2 基因突变检测结果及临床随访结果
病例4滤纸干血斑C5-OH初筛、血液复检结果持续升高(0.85 μmol/L、1.02 μmol/L),MCCC1基因存在致病突变(c.1518delG),确诊为MCCD。后续随访显示患儿生长发育正常(3岁1月),属于无症状MCCD患者,并且患儿携带β-地中海贫血CD41-42杂合突变。病例11携带2个MCCC2基因杂合突变(c.470A>G,c.1103G>A),突变等级为意义未明,最近一次患儿2岁6月时的随访显示生长发育正常,结合随访、血尿检测及基因结果诊断为无症状MCCD。病例5在半乳糖神经酰胺酶(galactocerebrosidase,GALC)基因上检测到c.1901T>C杂合突变,突变等级为致病变异,该基因为克拉伯病的致病基因,研究人员随访过程中未见异常。病例6 和8分别在MCCC1基因上检测到c.1331G>A和c.1732-1G>C杂合突变,突变等级为疑似致病,尿有机酸3-MCG和3-HIVA结果正常,随访未见患儿生长发育异常。病例7在半乳糖激酶1(galactokinase 1,GALK1)基因上发现2个意义未明的杂合突变;病例9在羟烷基辅酶A脱氢酶(hydroxyacyl-coA dehydrogenase,HADH)基因上发现c.112A>T 杂合突变,突变等级为意义未明,该基因突变可引起3-羟酰基辅酶A脱氢酶缺乏症或家族性高胰岛素性低血糖血症4型,随访过程中生长发育未见异常。
2.3C5-OH升高在多种疾病中的鉴别诊断流程 本研究中病例11孕期无特殊异常,足月剖宫产出生体重4.05 kg,身长50 cm,阿普加(Apgar)1 min和5 min评分均为10分,随访出生12 d、2月、3月生长发育检查无异常,最近一次在患儿年龄2岁6月时的随访中生长发育未见异常,患儿在初筛时C5-OH(9.24 μmol/L),复检升至C5-OH(11.61 μmol/L),临床提示MCCD、3-甲基戊烯二酰辅酶A水解酶缺乏症、3-羟基-3-甲基-戊二酰辅酶A裂解酶缺乏症、生物素酶缺乏症、全羧化酶合成酶缺乏症及β-酮硫解酶缺乏症等6种疾病可能,后续利用GC-MS检测尿有机酸结果提示3-羟基异戊酸、3-甲基巴豆酰甘氨酸升高,结合随访数据诊断为无症状MCCD患者。同时,在患儿MCCC2基因上发现2个意义未明的杂合突变,辅助临床进一步诊断。在参考美国相关流程图(American College of Medical Genetics, 2009)[11]的基础上,结合临床病例报道及国内专家共识和相关指南的建议[6,12-24],绘制了C5-OH升高可能的鉴别诊断流程,见图1。
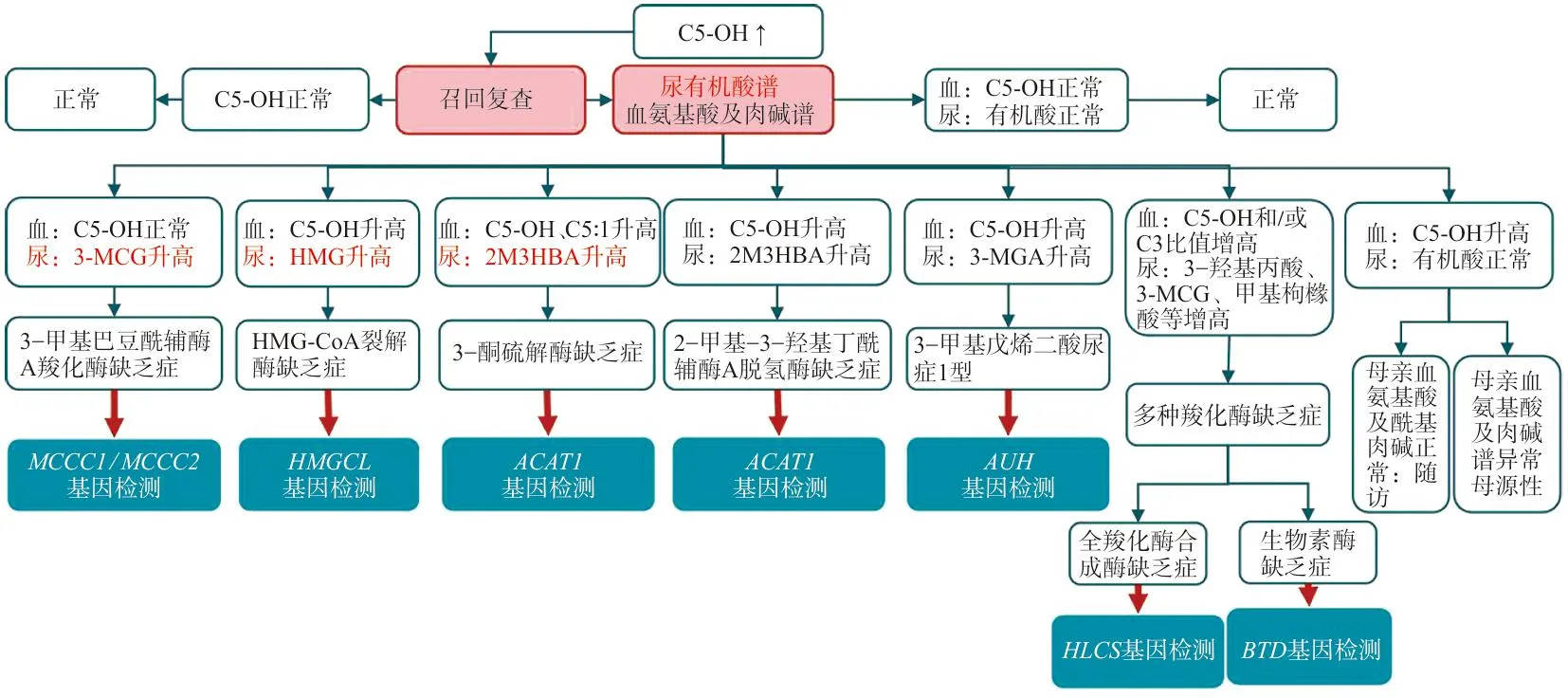
注:C5-OH,3-羟基异戊酰肉碱;C5:1,异戊烯酰肉碱;C3,丙酰肉碱;3-MCG,3-甲基巴豆酰甘氨酸;HMG,3-羟基3-甲基戊二酸;HMG-CoA,3-羟基-3-甲基戊二酰辅酶A;2M3HBA,2-甲基-3-羟基丁酸;3-MGA,3-甲基戊烯二酸。图1 C5-OH升高在多种疾病中的鉴别诊断流程
3 讨论
MCCD和多种羧化酶缺乏症是最常见的血液C5-OH增高的遗传代谢病,MCCD诊断的金标准是基因突变检测[22],本研究中病例4孕期无特殊异常,足月顺产出生体重2.55 kg,身长48 cm,阿普加(Apgar)1 min和5 min评分均为10分,最近一次在年龄3岁1月随访中生长发育未见异常;患儿初筛C5-OH为0.85 μmol/L,复检结果为1.02 μmol/L,提示可能为生物素缺乏、多种羧化酶缺乏症或MCCD,并建议尿液有机酸分析和基因检测,同时建议母亲进行串联质谱检测以排除母源性因素,但遗憾的是父母未能配合进行患儿尿液有机酸检测,患儿后期随访中进行基因突变检测,在MCCC1基因上发现2个杂合突变,其中1个突变等级为致病突变,另1个为意义未明,均提示MCCD。孟卫京等[22]在2019年报道1例MCCC基因上发现c.470A>G意义未明突变,但患儿尿中3-MCG轻度增高,临床无任何症状,6个月时检验各项指标均恢复正常;在另一篇关于MCCD的报道指出,所有患儿临床无症状,生长及智能发育正常,在21例疑似MCCD中仅11例检出MCCC1突变[23]。C5-OH异常增高在临床上还有一种原因是母源造成的一过性异常,2013年,宫丽霏等[24]在国内首报了5例母源性MCCD,在随访时均无临床症状,生长及智能发育正常。
MCCD患儿临床表型各异,大多数患儿为无症状的MCCD,少数可表现为神经系统发育受损[25],本研究中病例4和病例11患儿筛查时血液C5-OH浓度明显增高,召回后血浓度继续升高,且病例11伴尿3-MCG和3-HIVA含量显著增高,结合随访资料临床诊断为无症状MCCD患者;文献报道无症状MCCD患儿一般无需治疗,随访数年至10年仍无症状[26-27],但从临床的角度分析,本研究中的2例无症状MCCD患儿在后期随访过程中需密切关注患儿整体生长发育情况,预防患儿后期出现代谢异常而误诊为其他疾病,错过提前干预治疗的时间。
综上所述,本研究显示通过MS/MS、GC-MS、基因突变检测技术可实现C5-OH浓度异常增高的鉴别诊断,但本研究纳入的临床病例较少,研究结果仅能体现研究对象的个体情况,存在一定的局限性,对无症状MCCD患儿需要长期随访累积更多资料进行综合评估。
猜你喜欢
中老年保健(2022年4期)2022-08-22
今日农业(2021年16期)2021-11-26
家庭医学(下半月)(2019年9期)2019-10-12
猪业科学(2018年5期)2018-07-17
婚姻与家庭·性情读本(2017年3期)2017-04-27
中外医疗(2016年15期)2016-12-01
中外医疗(2015年11期)2016-01-04
健康必读(2014年2期)2014-06-23
中国中医药现代远程教育(2014年23期)2014-03-01
意林·少年版(2012年11期)2012-05-30
