
功能化多孔碳的制备及其对芳烃的吸附
2024-03-05 12:12:54肖杨韩亚婷熊鹤
中南民族大学学报(自然科学版) 2024年2期
肖杨,韩亚婷,熊鹤
(中南民族大学 a.化学与材料科学学院; b.催化材料科学国家民委-教育部重点实验室,武汉 430074)
对生物质多孔碳改性的方法有酸/碱改性法[1-2]、矿物改性法[3]、无机纳米材料改性法[4]和官能团改性法[5]等,通过增加生物碳吸附位点来提高吸附能力.如CHO 等[6]制备的氨基官能化单分散聚合物胶囊作为吸附剂,对水中的Cu2+、Cd2+、Zn2+离子的吸附能力分别可达476.71、205.01、262.6 mg·g-1.本文利用官能团改性法,对材料进行巯基官能团的修饰从而制备出功能化材料.巯基功能化主要有后接枝法和共聚法.在本文中使用的后接枝处理方法中,首先准备好载体,再通过含有巯基的化合物和载体之间的化学反应,将巯基化合物转化为载体;在共聚方法中,合成原料中带有功能性巯基的载体,使巯基能够稳定在基体中[7].本文对比了巯基功能化和生物碳在不同的外部物理条件(微波、超声波、蓝光)下生物碳的吸附效果,并结合SEM、Raman、FT-IR、XPS等分析多孔碳的形貌、官能团和元素构成,采用氮气吸脱附分析仪和相关计算模型获得比表面积和孔隙参数.
1 实验部分
1.1 试剂
300#液蜡油(其芳烃的主要成分是烷基苯和烷基萘,芳烃含量质量分数<3%)由江苏淮安清江石化提供,木质素磺酸钠和3-巯基丙基三甲氧基硅烷(MPTMS,97%)均购于上海阿拉丁试剂有限公司,氯化锌(ZnCl2,AR)、2,2,4-三甲基戊烷(异辛烷,AR)、液体石蜡(CAS: 8012-95-1,AR)和盐酸(HCl,AR)均购置于国药集团化学试剂有限公司.
1.2 样品表征
热重分析仪(TGA, TG209F3 Tarsus);傅里叶变换红外光谱仪(FT-IR, Nexus 470);X 光电子能谱(XPS, Multilab 2000);比表面积及孔径测量仪(V-Sorb 2800 P);X-射线粉末衍射仪(XRD,Bruker advance D8型);紫外可见分光光度计(Shimadzu UV-2550);高分辨场发射电子显微镜(SEM,Hitachi Su801);拉曼光谱(Raman,DXR).
1.3 样品的制备
1) 按1∶4的质量比准确称取木质素磺酸钠(SLS)和ZnCl2,与15 mL 的蒸馏水混合均匀,室温下经超声处理30 min,样品记为C.
2) 按1∶4 的质量比准确称取SLS 和ZnCl2,加入15 mL 的蒸馏水混合均匀,室温下蓝光照射并同时搅拌30 min,干燥后记为L.
3) 按1∶4 的质量比准确称取SLS 和ZnCl2,加入15 mL的蒸馏水混合均匀后放入微波炉中,700 W下加热30 min,样品记为W.
将SLS 和ZnCl2按1∶4 的质量比准确称量,与15 mL 的蒸馏水混合均匀,干燥后放入管式炉中,同时将样品C、L、W 放入管式炉中,在氮气保护下以5 ℃·min-1的升温速率从室温升至500 ℃,并保温2 h.冷却至室温后用质量比5%的盐酸磁力搅拌一定时间后抽滤,并用蒸馏水洗涤至中性,干燥后样品分别记为PSC和PSC-x(x代表C、L和W).
4) 取0.5 g 的PSC 样品分散于100 mL 的无水乙醇中,一边搅拌一边逐滴加入50 mL 的3-巯基丙基三甲氧基硅烷(MPTMS),在60 ℃下搅拌6 h后过滤,用乙醇反复洗涤后干燥,得到的样品记为PSC-SH.
1.4 吸附性能测试
分别称取吸附剂PSC-x和PSC-SH 各20 mg,放入装有50 mL 液蜡油且含萘浓度为3 g·L-1的三角烧瓶中,将烧瓶置于25 ℃恒温水浴振荡器中,以135 r·min-1的频率振荡,不同吸附时间下取样,进行离心得到上清液,用异辛烷稀释定容后测试吸附后的吸光度,每组测3 次取平均值,由公式(1)计算得萘的吸附量:
式中:C0和Ce分别为液蜡油中芳烃的初始浓度和吸附后的浓度,mg·L-1;qe为吸附量大小,mg·g-1;V为溶液的体积,L;m为吸附剂的质量,mg.
2 结果与讨论
2.1 形貌分析
对样品PSC 和PSC-x的形貌扫描结果如图1 所示.由图1 可见:PSC 表面分布着零星气孔且分布较为稀疏,孔径大多小于2 μm,而前期经超声振动和蓝光处理后再碳化得到的多孔碳样品在形貌上差别不大,PSC-C 和PSC-L 孔径较大且整个样品较为立体.相比之下,PSC-W 有更多的孔隙和更大的孔隙尺寸,PSC-SH 整个形貌与PSC 都不同.在接枝了巯基官能团后,样品呈现一种球状粘附现象,球体之间的孔隙也随着堆积的紧密程度而变化,增加了表面积和孔体积,巯基化的团聚增加了萘与吸附剂位点的接触概率.经不同方式处理的样品在孔隙上都有一定程度的改变,同时也为萘的吸附提供了吸附位点.从图1(f)PSC-SH 的高分辨透射扫描图可见:样品为圆形状实心颗粒堆叠,颗粒之间孔隙大小不一.

图1 样品的形貌扫描图和高分辨率透射电镜图Fig. 1 SEM and TEM images of the samples
2.2 FT-IR和XRD分析
红外光谱如图2 所示,其显示了所制备样品的表面官能团.不同外部物理条件(微波、超声波、蓝光)下三种吸附剂的官能团种类和PSC相同,这表明微波、超声波和蓝光处理并不会改变材料的表面官能团,在3432 cm-1处的峰对应水分子中的O—H 基本伸缩振动,2930 cm-1处的峰为—CH2的基本振动吸收,1620 cm-1为芳香骨架中的C=O 基团或C=C伸缩振动吸收[8],1025 cm-1和1380 cm-1处的峰分别为Si—O—Si 键或C—O 伸缩振动,613 cm-1处为烷基的C—O 基团振动.此外,在2400~2500 cm-1附近的—SH伸缩振动吸收峰不明显,这可能是由于红外光谱吸收峰较弱导致[9],结合后面的EDS 和XPS 分析,可以表明巯基已成功导入.
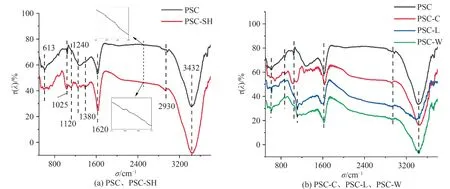
图2 样品的红外谱图Fig. 2 FT-IR spectra of the samples
对各样品的晶体结构特征采用XRD 进行表征,结果如图3 所示.由图3 可见:所有样品具有相同的峰型,位于2θ为24°和43.5°处的特征峰都较宽且弱,分别对应(002)和(100)晶格,且平面(002)峰较弱,表明样品接近理想石墨结构[10],样品的缺陷少,活性位点也较少.
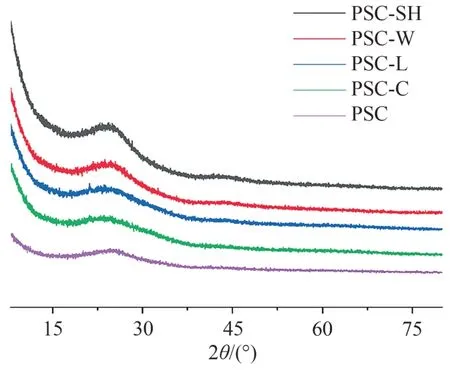
图3 五种样品的XRD谱图Fig. 3 XRD spectra of the five samples
2.3 拉曼和热重分析
拉曼光谱是表征碳纤维石墨化程度的一种有效工具,5 种样品的拉曼图谱如图4 所示,各样品在1324 cm-1(D 峰)和1576 cm-1(G 峰)波段出现两个最大强度的峰.D带与G带的强度比ID/IG可以用来衡量材料石墨化程度,较低的ID/IG值表示较高的石墨化程度[11]. PSC、PSC-C、PSC-L、PSC-W 和PSC-SH 样品的ID/IG值分别为0.78、0.80、0.88、0.86 和0.96,而PSC在5 种样品中的ID/IG值最低,表明PSC 的石墨化程度最高,活性位点最少.PSC-SH 的ID/IG值最高,说明活性基团巯基的引入降低了石墨化程度,活性位点增多,有利于萘的吸附[12].
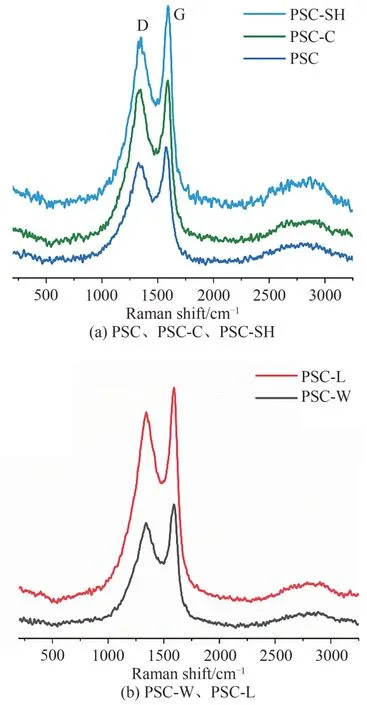
图4 样品的拉曼谱图Fig. 4 Raman spectra of the samples
木质素磺酸钠的热重分析如图5 所示.热重分析是在氮气环境下进行的,加热速度为10 ℃·min-1,温度范围为40~700 ℃.在0~200 ℃之间的损失主要发生在游离水的蒸发阶段,从DTG 曲线可以看出,在400~600 ℃范围内失重率急剧下降,这主要是由于材料在高温下发生裂解,并在600 ℃以后材料趋于稳定.
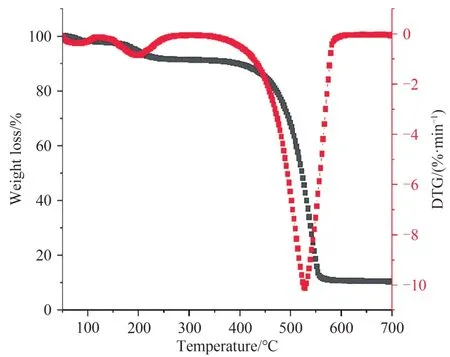
图5 木质素磺酸钠的热重分析谱图Fig. 5 TG spectrum of sodium lignosulfonate
2.4 EDS和XPS分析
样品PSC 和PSC-SH 的EDS 分析结果如图6 所示.由图6 可见:PSC 的主要元素组成为C、O 和S,与预期一致.而PSC-SH 的主要元素组成有C、O、Si和S,其中C、O、Si 和S 元素的原子百分比分别为83.665%、13.230%、0.690% 和2.415%,且Si 2p 的XPS 谱图如图6(c),结合能约为103.5 eV,Si 元素的存在也是成功引入MPTMS的侧面印证.
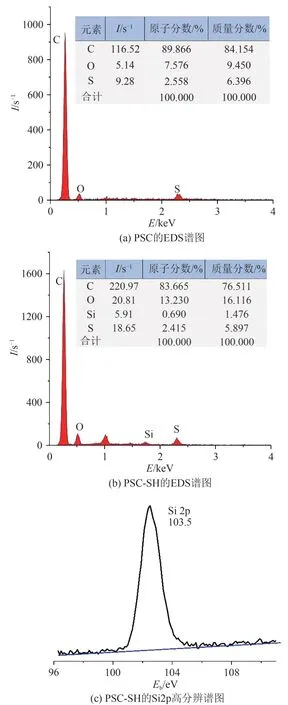
图6 EDS和Si2p高分辨谱图Fig. 6 EDS spectra and Si2p high resolution spectra of the samples
利用XPS 分析样品表面化学成分和元素组成,PSC 和PSC-SH 的XPS谱图如图7(a)所示,经能谱计算得的表面元素组成如表1 所示.XPS 全扫描图谱的结果与EDS 分析结果相互印证,再次证明巯基被成功引入.图7(b)和图7(c)显示了C1s 核心谱图可拟合4 个特征峰,分别位于284.7、285.3、286.4、289.1 eV,其中284.7eV 的峰归属于C=C/C—H 基团,285.3 eV 的峰归属于样品表面的碳碳基团(C—C),286.4 eV 的峰归属于PSC 样品结构中sp2杂化的碳(C—O),289.1 eV 的峰归属于sp2杂化的碳(O—C=O)[13].图7(f)为样品PSC-SH 的S 2p 谱图,显示为3 个特征峰(163.5、164.6、169.3 eV),分别归属于S—H、C—S 和O—S 基团[14-15],丰富的S 物种有利于萘在PSC-SH 表面的吸附.O 1s的高分辨光电子能谱如图7(d)和图7(e)所示分为531.2、532.8、533.6 eV三个特征峰,分别归属于C=O、C—O 和—OH基团[16].
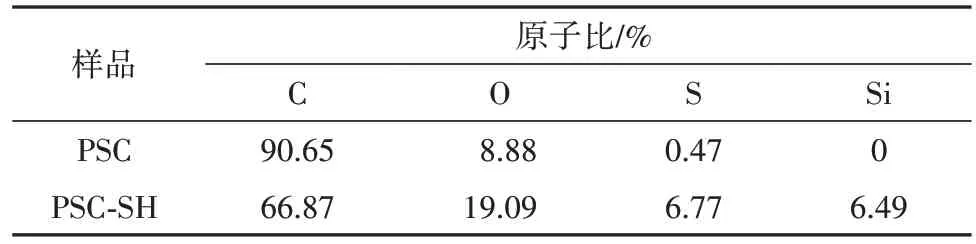
表1 PSC和PSC-SH的元素组成Tab. 1 Elemental composition of PSC and PSC-SH
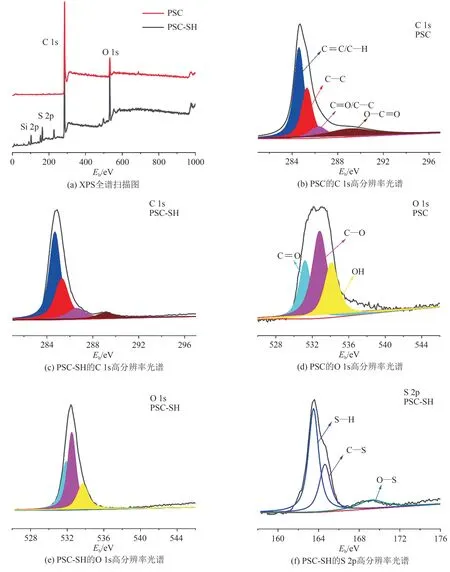
图7 样品的XPS谱图Fig. 7 XPS spectra of the samples
2.5 比表面积和孔结构分析
为了对材料的孔结构进行考察,分别对样品PSC、PSC-x 和PSC-SH 采用了氮气吸附-脱附测试,图8 显示了每个样品的吸附等温线和孔径分布曲线.由图8可见:所制备的样品的吸附等温线符合IV类吸附等温线.曲线显示[17],在p/p0较低时呈现急剧上升趋势,表明存在微孔结构;在较高压力范围内存在一定宽度的回滞环,表明存在一定数量的介孔.从图8(c)和图8(d)的孔径分布曲线可以看出,PSC和PSC-SH 的大部分孔道是微孔和介孔.样品PSCW、PSC-C 和PSC-L 的孔径大部分大于2 nm,少部分分布在1~2 nm 之间,说明材料主要是介孔为主,存在少量微孔.
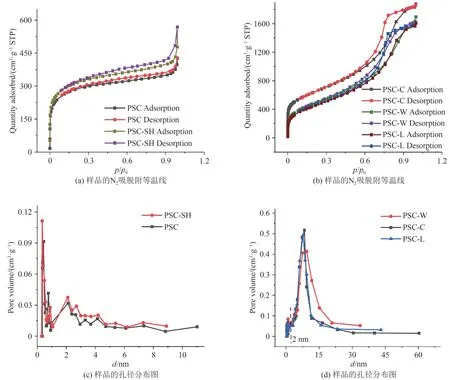
图8 样品的N2吸脱附等温线和孔径分布图Fig. 8 N2 adsorption and desorption isotherms and pore size distribution of samples
表2列出了各样品的孔结构参数,包括样品的比表面积(SBET)、孔容积(Vtot)、微孔体积(Vmic)以及平均孔径(Dav).可以看出:相比之下样品PSC和PSC-SH具有较低的比表面积,分别为1177.57和1152.48 m2·g-1,经不同外部物理条件(微波、超声波、蓝光)处理后表面积、孔容等有所增加.
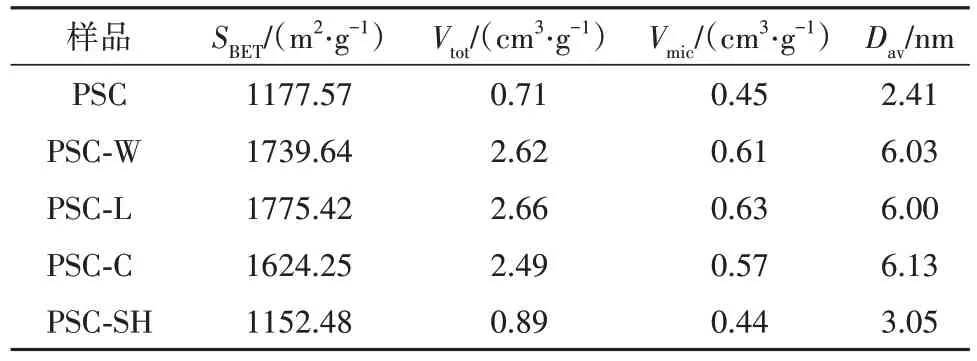
表2 样品的孔结构参数Tab. 2 Pore structure parameters of the samples
图9 为不同吸附材料对液体石蜡中萘的吸附量,随着改性条件不同,对萘的吸附量也高于PSC吸附剂.由图9 可见:PSC-SH 的吸附量最大,高达702.28 mg·g-1,尽管PSC-SH 由于团聚使得比表面积较小,其原因可能是巯基的引入堵塞了一些孔道,并在这些孔道上重新堆叠团聚,形成新的吸附位点,而萘的吸附主要通过引入的活性基团来实现的[12].而PSC-C(369.89 mg·g-1)吸附量只略高于PSC(339.65 mg·g-1),表明在超声辅助下物料混合更均匀,反应更彻底,符合上述材料比表面积的增加趋势.
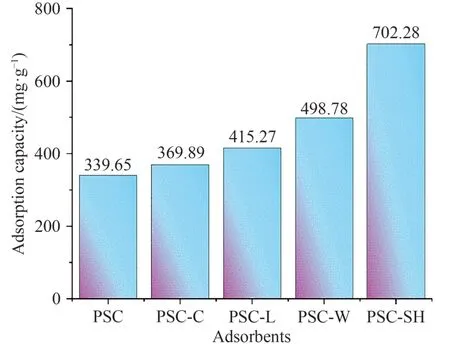
图9 PSC、PSC-C、PSC-L、PSC-W和PSC-SH对萘的吸附量Fig. 9 Adsorption of naphthalene by PSC, PSC-C, PSC-L, PSC-W and PSC-SH
2.6 吸附动力学
对五种吸附剂对萘的吸附动力学数据进行拟合,相关公式分别见式(2)和式(3).
如图10 为样品PSC、PSC-x和PSC-SH 的吸附动力学实验数据的拟合曲线,表3为动力学拟合参数,结果发现准二级动力学模型计算得到的平衡吸附量与实验得到的数据吻合良好,表明是在化学作用下的吸附过程.
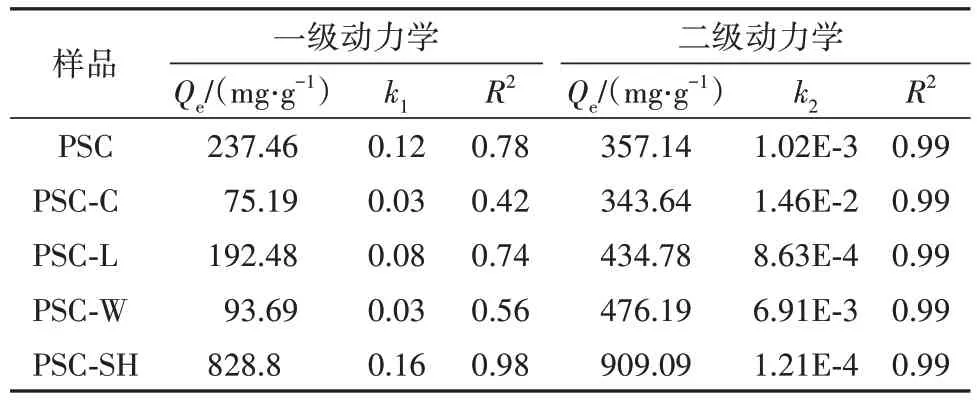
表3 动力学模型拟合参数Tab. 3 Kinetic model fitting parameters
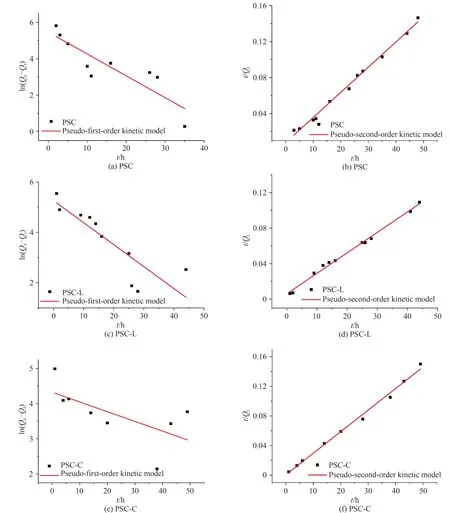
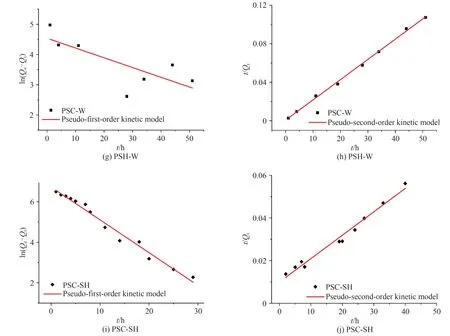
图10 样品的动力学拟合曲线Fig. 10 Kinetic fitting curves of the samples
分别采用Langmuir 和Freundlich 模型对萘的等温线拟合结果如图11所示,通过非线性回归分析方法得到了各模型参数的值(见表4).从表4的拟合数据结果可看出Langmuir 等温线模型的R2(0.92)大于Freundlich 等温模型的R2(0.84),且前者得到的最大吸附量与PSC-SH吸附平衡实验数据更加吻合.

表4 Langmuir 和Freundlich模型拟合参数Tab. 4 Langmuir and Freundlich model fitting parameters
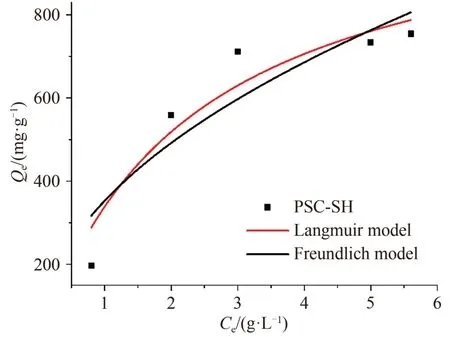
图11 PSC-SH等温吸附模型Fig. 11 PSC-SH isothermal adsorption modeling
3 结语
基于氯化锌对木质素磺酸钠的活化作用,经巯基改性和不同外部物理条件(微波、超声波、蓝光)处理得到的多孔碳.对液体石蜡中萘的吸附行为进行分析发现:这四种处理方法都会影响萘的吸附,相似的是对萘的吸附量均随时间而先增加后减少,最大吸附量达到702.28 mg·g-1,比表面积从1177.57 m2·g-1增加到1775.42 m2·g-1.结合SEM、Raman、FT-IR、比表面积和孔径分析结果发现:PSC-SH 与PSC 的比表面积和孔容上无明显差异,但PSC-SH 吸附萘的量是PSC的两倍,这可能是由于引入巯基,增加了样品的吸附位点,从而增加了吸附萘的量.动力学和热力学研究表明,PSC-SH的吸附过程是一种单分子层化学作用下的吸附.
猜你喜欢
中学生数理化·自主招生(2024年6期)2024-06-24 11:15:29
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21 07:51:12
铜仁学院学报(2018年6期)2018-07-05 09:47:34
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
材料科学与工程学报(2016年5期)2016-02-27 07:11:31
高中生学习·高二版(2015年3期)2015-05-21 15:54:27
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
四川师范大学学报(自然科学版)(2015年1期)2015-02-28 14:07:29
海洋科学进展(2015年1期)2015-02-27 13:16:16
无机化学学报(2014年8期)2014-02-28 17:32:48
