
高雄激素诱导多囊卵巢综合征表观遗传机制的研究进展*
2024-03-05 02:45梁梦梦张艳新邵欣欣郝文庆
中国病理生理杂志 2024年1期
梁梦梦, 赵 燕, 张艳新, 邵欣欣, 陈 聪△, 郝文庆
(1山东中医药大学药学院,山东 济南 250355;2济南市疾病预防控制中心,山东 济南 250021;3山东中医药大学中医文献与文化研究院,中医药经典理论教育部重点实验室,山东 济南 250355;4山东中医药大学第一临床医学院,山东 济南 250355;5巨野县中医医院,山东 巨野 274900)
多 囊卵巢综合征(polycystic ovary syndrome,PCOS)是一种以雄激素过多、排卵功能障碍和多囊卵巢为特征的代谢紊乱性生殖内分泌疾病[1]。根据全国临床大数据流行病调查显示,该病在育龄女性中的发病率为5.6%,其中无排卵性不孕的女性占75%,是引起育龄女性无排卵性不孕的主要原因[2]。当前,临床常以促排卵、降低血雄激素水平、缓解胰岛素抵抗等药物,以及手术进行治疗。
因PCOS 明显的异质性、遗传性,当前对其发病机制的研究尚未达成统一共识,认为主要与遗传、下丘脑-垂体-卵巢轴异常、胰岛素抵抗、雄激素过量等关系密切[3]。其中,表观遗传紊乱在该病的发生发展过程中发挥着核心作用,由宫内或产后不利环境引起的表观遗传变化对胎儿出生后的PCOS 样症状或相关临床改变具有引发作用[4]。高雄激素性妊娠是PCOS胎儿起源的主要因素,通过表观遗传突变导致子代青春期PCOS 的发病几率增加[5]。表观遗传是指DNA 序列没有发生改变,但是基因表达却发生了通过细胞有丝分裂或减数分裂可遗传的变化,并最终导致基因表型的改变[4],是连接早期生命特征及后期表型转变的重要机制,是临床PCOS女性及其子代疾病表征的重要遗传途径。因此,本文聚焦高雄激素诱导PCOS表观遗传机制的研究进展,以期为PCOS的治疗和诊断提供参考。
1 雄激素诱导PCOS模型及表观遗传机制的研究
PCOS 的发生伴随着的显著病理特征是雄激素升高,但是高雄激素与PCOS发病的具体相关性尚未明确,因而领域内专家提出了“雄激素致病”假说[6]。该假说指的是,幼儿过早暴露于高雄激素环境下,会使得成年以后的卵巢功能出现不同程度的PCOS 表征,这些表征是基因差异性表达的结果。可进行干预的常见雄激素有双氢睾酮(dihydrotestosterone,DHT)、脱氢表雄酮(dehydroepiandrosterone, DHEA)和丙酸睾酮(testosterone propionate, TP)。有报道表明,雄激素于产前或产后干预,通过招募不同的启动子和增强子调控DNA 表达,从而导致表观遗传改变及成年后出现病理改变[7],研究角度主要有组蛋白修饰、DNA 甲基化、非编码RNA 等(表1)。在动物模型中,猴子是反映疾病病理相对较为全面的物种,并且雌猴于自然状态下同样会发生高雄激素的症状[8]。小鼠模型可以为PCOS 提供分子层面的病理研究,如PCOS诱导的神经-内分泌改变。
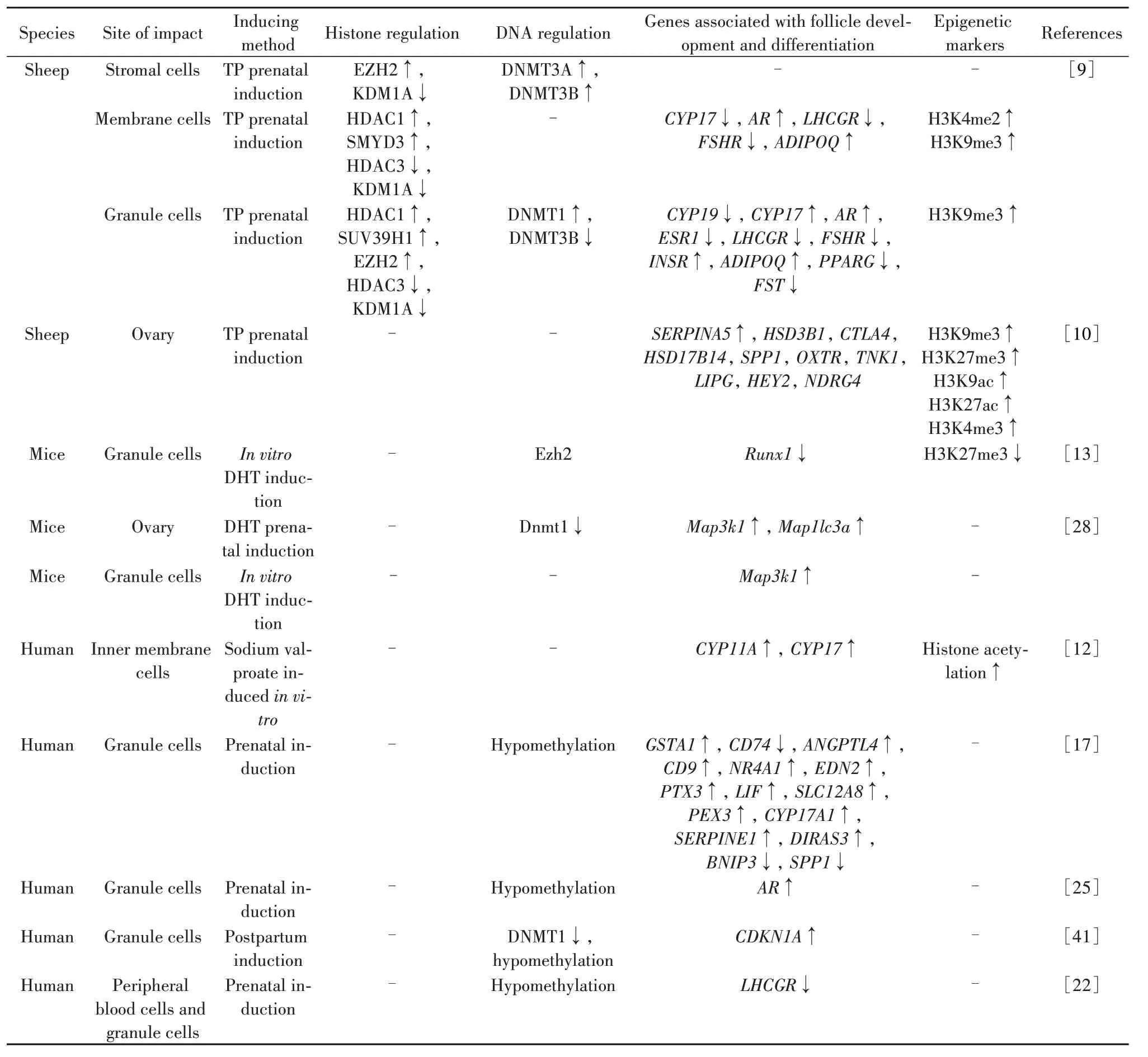
表1 雄激素诱导的PCOS模型表观遗传及相关基因Table 1. Epigenetic and related genes in androgen-induced PCOS models
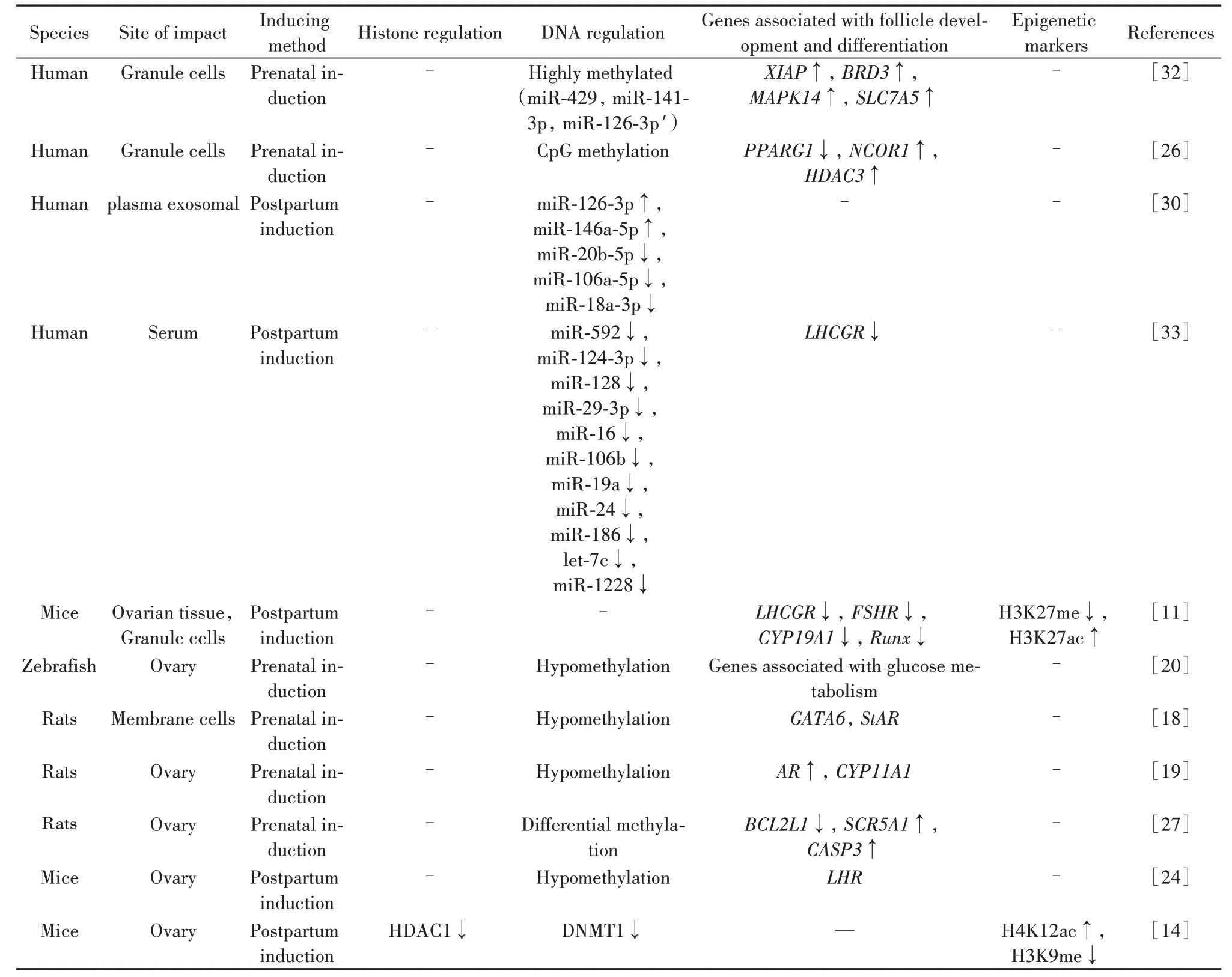
Table 1. (continued)
1.1 基于组蛋白修饰 组蛋白是一种八聚体结构,有组蛋白H2A、H2B、H3 和H4 各两个副本组成,由DNA 和连接蛋白H1 包裹。组蛋白尾部的化学修饰(如乙酰化、甲基化、磷酸化、泛素化和类泛素化)通过影响转录因子的结合从而影响基因转录和细胞表型(图1)。Guo等[9]和Sinha等[10]以绵羊为实验模型,于妊娠期30~90 d 腹腔注射TP,结果显示卵巢颗粒细胞中产前雄激素干预影响了DNA 甲基转移酶1(DNA methyltransferase 1,DNMT1)和DNMT3B的基因修饰,使得组蛋白H3 第9 位赖氨酸三甲基化(trimethylation of lysine 9 in histone H3, H3K9me3)水平升高,并显著影响了卵巢中膜细胞的组蛋白甲基化[9];对母羊和幼羊实施卵巢摘取并进行RNA 测序,显示母羊与幼羊之间的基因差异性表达并不一致,进一步开展母羊组蛋白研究,显示产前处理分离的卵巢中H3K9me3、组蛋白H3 第27 位赖氨酸乙酰化(acetylation of lysine 27 in histone H3, H3K27ac)和组蛋白H3 第9 位赖氨酸乙酰化(acetylation of lysine 9 in histone H3, H3K9ac)水平显著升高,但是组蛋白H3 第4 位赖氨酸三甲基化(trimethylation of lysine 4 in histone H3, H3K4me3)水平并无显著变化[10]。由此推测,产前雄激素的暴露会引起跨代表观遗传改变,这为PCOS的发生发展提供了一个研究方向。
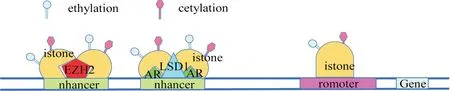
Figure 1. Epigenetic histone modifications in PCOS. EZH2: enhancer of zeste homolog 2; AR: androgen receptor; LSD1: lysinespecific demethylase 1.图1 PCOS中的表观遗传组蛋白修饰
产后雄激素诱导研究显示,雄激素可作用于基因的启动子以及增强子区域,通过减少组蛋白H3 第27 位赖氨酸三甲基化(trimethylation of lysine 27 in histone H3, H3K27me3),上调H3K27ac,调控下游多种基因的表达,促进PCOS 的发生发展[11]。Nelson-Degrave 等[12]通过临床检测38~40 岁患有PCOS 女性的卵巢内膜细胞与正常卵巢内膜细胞的雄激素生物合成水平,观察到丙戊酸钠可通过组蛋白乙酰化诱导雄激素的合成,从而造成卵巢多囊样改变。动物模型研究结果同样显示,组蛋白修饰的改变影响着PCOS 表征。小鼠卵巢颗粒细胞经25 nmol/L DHT 诱导后,PI3K/Akt通路快速磷酸化,从而抑制了zeste增强子同源物2(enhancer of zeste homolog 2, Ezh2)的活性,甚至可使Ezh2 的表达完全消失,雄激素的长期诱导使得黄体生成素(luteinizing hormone, LH)诱导的转录因子Runx1基因的启动子H3K27me3 抑制标记减少,引起了后期的排卵障碍[13]。小鼠卵巢卵母细胞荧光强度分析显示,DHEA 诱导处理的卵母细胞中DNMT1和组蛋白脱乙酰酶1(histone deacetylase 1,HDAC1)转录所需的mRNA 表达量下降,组蛋白H3第9位赖氨酸的去甲基化水平降低,组蛋白H4第12 位赖氨酸的乙酰化增加且与ROS 的产生呈正相关,表明ROS升高有可能是导致PCOS患者卵泡成熟率和排卵率低的原因[14]。
1.2 基于DNA 甲基化水平的修饰 DNA 甲基化是指在DNA 甲基转移酶的作用下,基因组胞嘧啶-磷酸-鸟嘌呤(cytosine-phosphoric acid-guanine, CpG)二核苷酸的胞嘧啶5 号碳位以共价键结合一个甲基基团(图2)。已有研究表明,PCOS 在子代具备显著的遗传性并伴随着遗传基因甲基化程度的改变[15-16],如类固醇相关合成基因[细胞色素P450家族17亚家族A(cytochrome P450 family 17 subfamily A,CYP17A)、CYP19A等]的甲基化,激素受体[黄体生成素/绒毛膜促性腺激素受体(luteinizing hormone/choriogonadotropin receptor,LHCGR)、雄激素受体(androgen receptor,AR)、卵泡刺激素受体(follicle-stimulating hormone receptor,FSHR)、胰岛素受体(insulin receptor,INSR)等]的甲基化及其他PCOS相关基因[丝裂原活化蛋白激酶激酶激酶1(mitogen-activated protein kinase kinase kinase 1,Map3k1)、微管相关蛋白1 轻链3α(microtubule-associated protein 1 light chain 3 alpha,Map1lc3a)等]的甲基化。
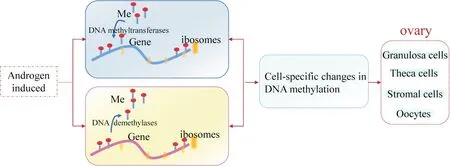
Figure 2. Regulation of epigenetic DNA methylation level in PCOS. Me: methyl group.图2 PCOS中表观遗传DNA甲基化水平的调控
临床对22~36 岁110 名患有PCOS 的妇女以及119 名排卵周期正常的体外受精女性的卵巢颗粒细胞进行RNA 测序分析,结果显示卵巢颗粒细胞中脂类和类固醇激素合成基因上调,结合DNA 定量甲基化分析,显示脂类和类固醇类相关基因启动子呈现低甲基化,这可能是导致相关基因异常表达的原因[17]。已有研究表明,在游离睾酮诱导的大鼠卵巢膜细胞中,类固醇相关基因CYP17、GATA结合蛋白6(GATA binding protein 6,GATA6)和类固醇合成急性调节蛋白(steroidogenic acute regulatory protein,StAR)的启动子甲基化水平降低,其中GATA6和StAR启动子-520(CpG9)和-822(CpG4)位点的甲基化显著降低[18]。检测PCOS 大鼠模型外周血和卵巢组织中AR、CYP11A、CYP11A1、CYP17A和CYP17A1的甲基化状态,结果显示在外周血细胞中AR出现3 个低甲基化位点,CYP11A1出现1 个低甲基化位点,在卵巢组织中AR出现5 个低甲基化位点,CYP11A1出现1个低甲基化位点;此外,组织特异性甲基化分析显示,与卵巢和血液相比,AR和CYP11A1在睾丸中均表现出显著的低甲基化[19]。以上研究提示,产前雄激素诱导类固醇合成相关基因低甲基化是导致PCOS 伴高雄激素血症的可能原因之一。除此以外,科研人员以过度暴露于DHT环境下的成年斑马鱼的子一代受精卵为研究对象,发现子一代斑马鱼整体出现低甲基化现象,葡萄糖耐受实验显示,子一代斑马鱼的空腹血糖及餐后血糖均出现升高[20]。这提示产前雄激素暴露可能导致卵巢表观基因组和葡萄糖稳态的跨代改变,但二者之间的关系尚未明确。
伴随激素水平变化的激素受体也会发生相应的甲基化水平变化。全基因关联分析结果显示,LHCGR是PCOS 的易感基因[21]。为探讨LHCGR表观遗传成分致PCOS 发病的机制,Wang 等[22]对采集的PCOS 女性外周血细胞和颗粒细胞的DNA 甲基化进行分析,结果显示LHCGR启动子区域CpG 出现低甲基化状态;临床免疫组化结果显示,PCOS 患者的LHCGR 水平比正常组高,同时伴有CYP17A1、3β-羟基类固醇脱氢酶II(3β-hydroxysteroid dehydrogenase II, 3βHSDII)和17β-羟基类固醇脱氢酶(17β-hydroxysteroid dehydrogenase, 17βHSD)的高表达[23];DHEA 诱导的小鼠卵巢出现LHCGR低甲基化的情况[24],这与临床结论一致,提示LHCGR低甲基化是PCOS 发病的潜在机制。在PCOS 相关度较高的基因AR的DNA 甲基化程度的研究中,Desmawati 等[25]比对PCOS 女性和正常女性颗粒细胞中的DNA 甲基化测序结果,结果显示PCOS 女性颗粒细胞中AR基因启动子的DNA 甲基化水平较低,提示颗粒细胞中DNA低甲基化会增加AR水平,导致高雄激素诱导的PCOS的发生。
上述研究表明,PCOS 的发病与激素及其受体合成基因的甲基化程度有关。为探究高雄激素诱导的非激素类基因的表观遗传变化,及其与PCOS卵巢功能障碍间的关系,Qu 等[26]对高雄激素血症和非高雄激素血症PCOS 女性卵巢颗粒细胞进行基因甲基化测序,结果显示过氧化物酶体增殖物激活受体γ1(peroxisome proliferator-activated receptor gamma 1,PPARG1)启动子中的2 个高甲基化CpG 位点和核受体辅阻遏物1(nuclear receptor corepressor 1,NCOR1)启动子中的5 个低甲基化CpG 位点仅见于高雄激素血症PCOS女性。Zhang等[27]通过建立产前雄激素化雌性大鼠模型,观察其卵巢形态,表现为闭锁卵泡和囊性卵泡增多,甲基化检测结果显示基因BCL2L1和SCR5A1出现高度甲基化,并伴有血清葡萄糖、17-羟孕酮和雄激素水平的升高。产前DHT诱导高雄激素小鼠模型显示,卵巢DNMT1 表达水平显著下降,使得自噬相关调控基因Map3k1和Map1lc3a甲基化水平显著降低,从而促进卵巢颗粒细胞自噬水平升高,引发卵泡发育障碍[28]。以上研究提示,Map3k1、Map1lc3a、PPARG1、NCOR1、BCL2L1、SCR5A1等非激素类基因的甲基化变化参与高雄激素诱发PCOS 的卵巢功能障碍,其潜在作用机制可能与高雄激素环境对机体葡萄糖稳态的影响有关。
1.3 基于非编码RNA的调节
1.3.1 基于微小RNA(microRNA, miRNA)水平的调节 miRNA 是一类内源性非编码的、长度约为21~25 个核苷酸的小RNA,具有高度的保守性、时序性、组织特异性,以及广泛的基因调控作用,主要通过调控靶基因的转录以调控蛋白质的表达,从而改变生物的性状,因此维持miRNA 的稳定对于维持细胞的正常活动十分重要[29]。已有研究表明,PCOS 患者内分泌紊乱、卵泡闭锁和卵泡发育不全等症状受miRNA基因调控作用的影响[30],如miRNA-21是在正常卵泡发育过程中由LH 诱导的高表达miRNA 之一,高雄激素诱导下的颗粒细胞中miRNA-21水平骤降,可引发细胞凋亡[31];PCOS 患者卵巢颗粒细胞表达的830 个基因和30 个miRNA 中,7 个miRNA 负调控靶mRNA 表达,130 个miRNA 启动子显著不同甲基 化,其 中miRNA-429、miRNA-141-3p 和miRNA-126-3p 的启动子高甲基化受miRNA 表达抑制影响,进而导致相应基因XIAP、BRD3、MAPK14和SLC7A5上调[32]。高雄激素诱导DNA 甲基化水平改变对PCOS 发生发展影响的研究已表明,高雄激素血症PCOS女性卵巢颗粒细胞中PPARG1启动子高甲基化且NCOR1启动子低甲基化,经研究,造成基因甲基化改变的主要原因是PPARG1mRNA表达量降低,而NCOR1mRNA 表达量增高[26]。LHCGR是PCOS 的易感基因,其低甲基化是PCOS 发病的潜在机制,进一步研究显示PCOS 患者体内游离的miR-592 水平低于正常组,且miR-592 与LHCGR基因的3'非翻译区发生相互作用从而下调LHCGR 蛋白的表达[33]。这些研究揭示高雄激素环境通过影响相应miRNA 对其靶基因的转录调控,进而影响PCOS的发生发展。
1.3.2 基于长链非编码RNA(long noncoding RNA,lncRNA)水平的调节 lncRNA 是长度大于200 nt 的RNA 分子,通常认为他们并不编码蛋白,而是以RNA 的形式在多种层面上(表观遗传调控、转录调控、转录后调控等)参与蛋白编码基因的调控。Jin等[34]比对PCOS 患者与正常组卵巢颗粒细胞lncRNA测序结果,显示有1 361 个差异lncRNA;Wang 等[35]比对卵泡液内lncRNA 测序结果,显示有1 253 个上调lncRNA,613 个下调lncRNA。这表明lncRNA 可能在PCOS发病机制中发挥重要作用,并有可能成为PCOS疾病诊断和治疗的靶标。
研究显示,lncRNA 可以通过调控转录因子实现对miRNA 的调节以及对RNA 的编辑和降解[36-37]。PCOS 患者卵巢颗粒细胞中高表达的lncRNA H19 可降低miRNA-19b 的表达,且萤光素酶报告基因检测证明lncRNA H19 可以与miRNA-19b 结合而调节下游靶点,从而起到对卵巢颗粒细胞的调控作用[38]。lncRNA ZFAS1 可与miRNA-129 结合而促进HMGB1表达,导致PCOS 患者卵巢颗粒细胞的内分泌紊乱,促进多囊卵巢颗粒细胞的凋亡[39]。颗粒黄体细胞中miRNA-145 和miRNA-182 的相对表达显著下降,卵泡液内miRNA-182则差异上调,且与PCOS患者的激素水平呈显著相关性[40]。这些研究表明,lncRNA 通过调节miRNA 参与PCOS 的发病机制,且在激素的合成和代谢中发挥着重要的作用。
lncRNA 也可以通过调控DNA 甲基化影响PCOS的发生发展。lnc-MAP3K13-7:1 作为PCOS 患者颗粒细胞高表达的lncRNA,可诱导泛素化导致DNMT1降解,抑制DNMT1 的表达,从而导致DNMT1 依赖的细胞周期蛋白依赖性激酶抑制物1A(cyclin-dependent kinase inhibitor 1A,CDKN1A)启动子低甲基化,增加了CDKN1A的表达,导致颗粒细胞生长减弱[41]。
2 结语与展望
PCOS 严重影响着育龄妇女的健康,是引起育龄女性无排卵性不孕的主要原因,是影响国计民生的重大医疗难题。目前,国际多以Rotterdam 标准、NIH标准和AES 标准进行诊断,但是在不同地区和不同专科中的应用情况存在较大差异[42]。由于PCOS 复杂多样的临床表现,导致国内外在诊疗PCOS时存在诊断延迟、治疗效果不佳等问题[43-44]。因此,PCOS早期疾病临床诊断标志物的鉴定及病理机制的深入研究能够为攻克PCOS提供有力支持。
雄激素诱导的表观遗传是当今诊治PCOS 的研究热点,现有研究深化了对PCOS 发病机制的认识。但是综合以上,现阶段的研究仍存在一定的问题:研究成果呈现了很多内源性的改变,但是环境因素诱导的这些改变究竟是如何调控疾病发生及演变的过程和机理尚未阐明;研究样本差异大、实验可重复性低、涉及基因面广、研究不够深入,导致目前仍未筛选出对PCOS发病起到决定性影响的基因;动物研究证实产前雄激素暴露会引起跨代表观遗传改变,但是动物与人类之间始终存在着差异,当前对人类母代、子代的研究较少,跨代分离研究并不深入。
由于PCOS具有明显的遗传性和异质性,表明最少是由两对以上致病基因的遗传信息累积导致,并不遵从孟德尔遗传规律,因此应该深化分子遗传学研究,采用单核苷酸多态性筛查基因型与疾病的关联性,并进行单体型研究和家系分析;同时,分析相关基因的功能,明确调控机制,以筛选出PCOS 的诊断生物标志物和治疗靶标,明确治疗方向。
猜你喜欢
现代仪器与医疗(2021年6期)2022-01-18
河北果树(2021年4期)2021-12-02
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国计划生育和妇产科(2021年9期)2021-04-18
上海公路(2019年3期)2019-11-25
中国临床医学影像杂志(2019年6期)2019-08-27
福建基础教育研究(2019年10期)2019-05-28
中成药(2017年9期)2017-12-19
川北医学院学报(2015年5期)2015-12-05
郑州大学学报(医学版)(2015年2期)2015-02-27
