
索非那新琥珀酸盐合成工艺比较
2024-02-29 07:36毛波
工业微生物 2024年1期
毛 波
广西奕安泰药业有限公司,贵港 广西 450800
琥珀酸索非那新(solifenacin succinate),化学名为(3R)-1-氮杂双环[2.2.2]辛烷-8-基-(1S)-1-苯基-3,4-二氢-1H-异喹啉-2-甲酸酯丁二酸盐,是日本Astellas 公司开发的选择性毒蕈碱M3 受体拮抗剂,能选择性松弛膀胱逼尿肌,用于治疗有尿频尿急症状的膀胱多动症(OAB)[1]。

图1 琥珀酸索非那新
目前,公开报道的合成1 的方法有多种,在生产中具有价值的主流路线均是以(S)-1-苯基-1,2,3,4-四氢异喹啉(2)和(R)-3-奎宁环醇(3)作为关键中间体。其中,合成方法上的主要差别在于对2 的转化方式(图2)。路线A 中2 与氯甲酸乙酯缩合,然后在强碱NaH 作用下,在甲苯中与(R)-3-奎宁环醇(3)回流,发生酯交换反应生成6。路线B中先将3 氯甲酰化,然后其盐酸盐与2 在吡啶溶剂中反应生成6。路线C 是通过对2 引入离去基团(Lv)后,再与3 进行缩合得到6。上述方法各有优劣势。例如,路线B 使用了大量的吡啶,按目前流行的绿色化学的观点来看,环保安全不能满足要求。

图2 三种路线的转化方式
1 实验部分
1.1 路线A
1.1.1 化合物4((S)-1-苯基-3,4-二氢异喹啉-2(1H)-羧酸乙酯)的合成
将甲苯溶剂和水的混合溶剂(甲苯和水的量分别为12 mL 和8 mL)开启搅拌,在搅拌的过程中分别加入化合物2((S)-1-苯基-1,2,3,4-四氢异喹啉)(2.4 g,5.7 mmol)和无水碳酸钾(1.66 g,6.0 mmol);搅拌均匀后,将整个体系降温到0~10 ℃之间,然后开始滴加氯甲酸乙酯(1.3 g,6.0 mmol),在滴加过程中要保持整个体系的温度维持在0~10 ℃之间;滴加完毕后,撤掉冷媒,将体系内的温度自然回温至室温(25~30 ℃),并在室温下反应1 h。反应完毕后,分液,弃掉水层,将有机层进行4 次洗涤、分液,洗涤剂分别为自来水(10 mL)、盐酸(1 mol/L,4 mL)、自来水(10 mL)和饱和氯化钠溶液(16 mL);洗涤完成后,还需要进行干燥处理(无水硫酸钠),然后将滤液控制在35 ℃温度下进行减压蒸馏,蒸至无液体流出,得到无色油状物4(3 g)。
1.1.2 化合物6(索非那新)的合成
在甲苯溶剂(30 mL)中加入上一步制备的化合物4(3 g,5.3 mmol),开启搅拌,将甲苯溶液逐渐加热至110~120 ℃,并在此温度下搅拌2 h,以除掉溶液中的水分;然后冷却降温到70~80 ℃,加入(R)-3-奎宁环醇(4.04 g,12.2 mmol),再分批次地加入60%氢化钠(0.42 g,8.8 mmol)。加入完毕后,将整个甲苯体系升温至140 ℃,形成微回流,回流的液体(乙醇副产物)分开收集,保持此反应状态6 h。停止保温,并将反应体系自然冷却至室温(25~30 ℃),将溶液进行3 次洗涤、分液,洗涤剂分别为饱和氯化钠溶液(20 mL)、自来水(16 mL)、盐酸(1 mol/L ,8 mL);最后再用2 mol/L 氢氧化钠溶液将有机相调至碱性pH 约为10。接着再用乙酸乙酯(40 mL)萃取,并依次用水(16 mL)和饱和氯化钠溶液(20 mL)洗涤。最后经无水硫酸钠干燥、过滤,滤液在35 ℃以下减压蒸馏至无液体流出,得淡黄色油状物6(3.38 g,以6 计收率81.3%)[2],直接投入下步反应。
1.1.3 琥珀酸索非那新的合成
将Step 2 的黄色油状产物1.69 g 溶于15.8 mL乙酸乙酯和0.7 mL 无水乙醇混合溶液中,加入0.54 g 琥珀酸,加热回流搅拌反应20 min,加热后很快有大量白色晶体析出。体系搅拌冷却至室温(25~30℃),搅拌析晶2 h,然后冰浴中降温至10 ℃搅拌析晶1 h。抽滤,晶体用10 ℃的乙酸乙酯溶剂洗涤(500 mL×2)。60 ℃真空干燥得粉末状白色晶体,干重:1.63 g,总收率75%。纯度99.3%(HPLC 归一化法:色谱柱Phenomenex Gemini C18柱(4.6 mm×250 mm,5 μm);流动相:磷酸氢二钾溶液(磷酸氢二钾4.35 g 溶于水500 mL 中,用磷酸调至约pH 6)-乙腈(55∶45);检测波长210 nm;流速1 mL/min;柱温40℃;tR 5.7 min[3]。
1.2 路线B
1.2.1 化合物7 的合成
氮气保护下,将120 g Lv 酰化剂(结构如图3)加入800 mL 甲苯中,密封反应容器,室温下搅拌溶解,冰浴将反应体系冷却至3 ℃。200 g 化合物2,加入600 mL 甲苯,室温下搅拌溶解(溶解过程为吸热反应,可适当加热促进溶解);溶解完全后加入15 g(77 mL)吡啶,冷却至室温,缓慢滴加至上述体系中,保持温度低于10 ℃。反应放热很剧烈,前期滴加应缓慢,中期可稍快,后期可快速滴加。滴加完毕后移去冰浴,加热至80 ℃搅拌反应45 min(升温至50℃开始计时),TLC 监测反应结束,体系冷却至室温。加入60 g 硅藻土,搅拌10 min,抽滤除去吡啶盐酸盐,得黄色澄清溶液;60~65 ℃减压蒸除溶剂,得红褐色油状物298.86 g。
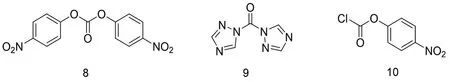
图3 路线C 中不同Lv 酰化试剂
1.2.2 化合物6 的合成
110.48 g 化合物3、THF 1 600 mL 加入5 000 mL 反应瓶中。先缓慢加入60% NaH 至体系无明显气饱溢出,再投入共计86.88 g 的60% NaH。加热至回流,搅拌45 min。Step1 油状产物298.86 g 加入500 mL THF,搅拌溶解;缓慢滴加到上述回流体系中,滴加速度约为3~4 滴/s,总滴加时间约为2.5 h。TLC 跟踪反应,至化合物3 点基本消失时结束反应。体系冷却至室温(30 ℃),缓慢滴加水,完全淬灭未反应的NaH,用水(800 mL)洗涤5 min;分液,水相加入500 mL 甲苯搅拌萃取5 min;分液,合并有机相,加入饱和NaCl 溶液800 mL 洗涤5 min;分液,有机相用20% HCl 500 mL 提取20 min;分液,水相用2 mol/L NaOH 溶液调节pH 约为10;水相用1 600 mL 乙酸乙酯提取10 min;分液,有机相用饱和NaCl 溶液800 mL 洗涤5 min;分液,有机相加入500 g 无水硫酸钠搅拌干燥4 h,50~60 ℃减压旋蒸除去溶剂,得黄色油状产物326.06 g。
1.2.3 琥珀酸索非那新的合成
将Step 2 的黄色油状产物326.06 g 溶于1800 mL EA 和135 mL 无水乙醇混合溶液中,加入102.57 g 琥珀酸;加热回流搅拌反应20 min,很快有大量白色晶体析出。体系搅拌冷却至室温(30 ℃),搅拌析晶2 h;然后冰浴中降温至10 ℃搅拌析晶1 h。抽滤,晶体用10 ℃的EA 洗涤(500 mL×2)。60 ℃真空干燥得粉末状白色晶体,干重:379 g,总收率90.8%。
2 结果与讨论
将上面两个实验的过程和结果进行对比可发现,Path A 的合成路线先由(S)-1-苯基-1,2,3,4-四氢异喹啉与氯甲酸乙酯缩合生成酯类化合物,然后再与(R)-3-奎宁环醇发生酯交换反应,成盐,总收率约75%;其中第一步反应简单高效,但在第二步的酯交换反应中,需要高温并移走产生的乙醇才能推动反应平衡右移,长时间的高温使副反应加剧,杂质增多,给产物的纯化带来不利影响。如应用到中试或生产,偏高的能耗也是一大弊端。Path B 的合成是由(S)-1-苯基-1,2,3,4-四氢异喹啉与Lv 酰化试剂发生酰化反应生成甲酰化物质后,再与(R)-3-奎宁环醇发生酯化反应,成盐,总收率约90%;其收率比较客观,纯度也比较高,工艺快捷安全,生产效率高,操作的可行性和安全性比Path A 高。
3 结论
通过两条路线的对比发现,路线B 不仅具有较高的收率,而且产品质量好、工艺快捷安全、生产效率高,更适合用于工业化生产。
猜你喜欢
净水技术(2022年11期)2022-11-10
中国民间疗法(2021年18期)2021-11-02
中国纤检(2021年9期)2021-09-30
中国特种设备安全(2021年2期)2021-07-21
中华养生保健(2020年9期)2021-01-18
无机化学学报(2019年2期)2019-02-27
浙江工业大学学报(2017年5期)2018-01-22
当代化工(2016年9期)2016-10-28
国外医药(抗生素分册)(2016年3期)2016-07-12
中国洗涤用品工业(2015年9期)2015-02-28
