
增材制造医疗器械行业的发展现状
2024-02-29 06:28:00朱超挺通信作者周天绮胡彬
医疗装备 2024年2期
朱超挺(通信作者),周天绮,胡彬
浙江药科职业大学医疗器械学院 (浙江宁波 315100)
增材制造(additive manufacturing,AM)又名3D打印,于1988 年由麻省理工ElySachs 教授首次给出命名,主要被用于满足模型制作和快速成型的高度专业化需求,现已成为计算机辅助设计(computer aided design,CAD)和快速制造的通用技术平台[1]。AM 技术以“逐层叠加”为核心思想,以三维模型为样本,实现了从无到有的加工过程。与传统的减材制造和模具注射成型技术相比,虽然AM 技术在加工速度和质量上存在缺陷,但其改变了传统的制造理念与方式,在制造领域拥有设计灵活性高、个性化成本低、原材料利用率高及工序少等独一无二的优势。
AM 医疗器械是按仿生形态学、生物体功能及微环境等要求,用AM 技术制造的模拟人体体内生物学结构和功能的医疗器械[2]。AM 医疗器械呈现出多领域学科交叉的技术特点:覆盖从临床采集患者数据到制造、再到临床的过程,包括影像获取、产品设计、格式转换、材料控制、打印加工、后处理等环节,每个环节都对最终产品具有决定性影响[3]。以上特点也对产品的全生命周期质量控制提出了严苛的监管要求。
随着AM 技术在齿科和植入器械领域的快速发展,监管部门亟须制定更多的该领域国家标准与行业标准以响应市场需求。当前,AM 医疗器械标准体系及监管制度尚未建立完善,这必将对AM 医疗器械的全生命周期质量控制提出挑战。因此,本研究对AM 医疗器械行业发展现状进行综述,并提出了相关建设思路,以期系统化推进AM 产业在医疗领域的高质量快速应用。
1 国内已取得AM 医疗器械注册证现状
医疗器械按风险等级可划分为一类医疗器械(低风险)、二类医疗器械(中风险)和三类医疗器械(高风险)。截至2023 年7 月23 日,国内已获批的AM 医疗器械注册证共计76 个,其中一类7 个(占比9.21 %)、二类55 个(占比72.37 %,含1 个进口许可产品)、三类14 个(占比18.42 %,含2 个进口注册产品),见图1。从图1 可以发现,二类产品占据已注册产品的50 %以上,且三类产品占比远高于一类产品。这表明,目前AM 医疗器械产品主要方向为二类和三类产品。图2 为2017—2023 年注册及备案的AM 医疗器械数量变化趋势。从图2 可以发现,2017—2019 年,AM 医疗器械注册增速较为缓慢;从2020 年开始AM 医疗器械注册数量开始迅速增加,年增长率高达60 %以上,展示了这一领域的良好前景。

图1 截至2023 年7 月23 日国内累计获批的AM 医疗器械数量

图2 2017—2023 年的历年数量
图3 为注册AM 医疗器械产品的省份分布图。由图可知,重庆、北京和上海占据前3 位,其数量总和接近所有已注册产品的47.37 %。其中,三类产品主要分布在北京(8 个境内三类注册证)和上海(2 个进口三类注册证),分别占三类产品总数的61.54 %和15.38%。
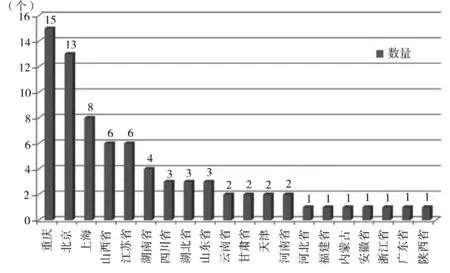
图3 截至2023 年7 月23 日国内累计获批的AM 医疗器械省份分布情况
表1 为截至2023 年7 月23 日国内累计获批的部分AM 医疗器械。由表可知,一类产品主要以工辅具、隔离眼罩和AM 耗材为主,二类产品主要以手术导板和器官模型为主,三类产品以植入物为主。

表1 截至2023 年7 月23 日国内累计获批的部分AM 医疗器械
2 国内外标准化组织及AM 标准现状
2.1 国外标准化组织及AM 标准现状
针对AM 的标准发布,国外以国际标准化组织TC 261 技术委员会(Technical Committee TC 261 of the International Organization for Standardization,ISO/TC 261)和美国材料与试验协会F42委员会(American Society for Testing and Materials F42 Committee,ASTM F42)为主要起草单位。两者联合制定了TC 261/184/156 等系列标准,为AM 的标准化奠定了基础。其他组织包括美国国防部(United States Department of Defense,DOD)、美国联邦航空管理局(Federal Aviation Administration,FAA)、欧洲航空安全局(European Aviation Safety Agency,EASA)等。
目前,已有的国外AM 标准包括ISO-17296 系列标准、F42 系列标准及ISO/ASTM 联合发布的系列标准。基本涵盖了术语、原材料、工艺、数据和试验方法等内容,以满足AM 工业要求。
2.2 国内标准化组织及AM 标准现状
2015 年4 月,全国AM 标准化技术委员会(SAC/TC 562)成立,负责AM 术语和定义、工艺方法、测试方法、质量评价、软件系统及相关技术服务等领域国家标准的制修订工作[4]。对口ISO/TC 261,秘书处由中机生产力促进中心承担,由中国机械工业联合会负责日常管理和业务指导。目前,全国AM 标准化技术委员会的标准化活动主要围绕机械工业和航天领域,已组织制定16 个国家标准,但是缺少医疗技术及产品相关标准。
2019 年1 月22 日,国家标准化管理委员会印发公告(2019 年第2 号)[5],正式成立全国AM 标准化技术委员会测试方法分技术委员会(SAC/TC 562/SC1),主要负责AM 领域的专用材料、设备及成形件质量及安全测试方法等国家标准的制修订工作。
2019 年11 月,中国食品药品检定研究院作为全国医用AM 技术标准化技术归口单位获得国家药品监督管理局批准,开展对AM 医疗器械产品质量控制技术的研究,推进国内医用AM 技术标准化建设。截至2023 年7 月23 日,国内已发布医用AM国家标准及行业标准共计5 项,见表2。其中3 项行业标准归口单位为中国食品药品检定研究院,分 别 为YY/T 1701-2020、YY/T 1809-2021、YY/T 1851-2022。

表2 截至2023 年7 月23 日国内现行医用AM 标准
由于国家标准和行业标准更关注基础性、通用性、涉及安全性指标的标准研究,因此制定周期较长[6]。而团体标准则具有快、新的特点。AM 领域的团体标准制定非常活跃(已发布38 项团体标准),如中国医疗器械行业协会3D 打印医疗器械专业委员会共发布团体标准30 项,涵盖原材料、设备、软件、产品、方法、质量体系等,产品类别涉及骨科、口腔领域。目前已制定医用AM 团体标准的单位包括中国医疗器械行业协会3D 专委会、中国生物医学工程学会、中国生物材料学会等。
3 国内AM 医疗器械的监管发展现状
2014 年,AM 骨科植入物被列入《需进行临床试验审批的第三类医疗器械目录》[7]。2015 年,3D 打印手术导板或手术演练模型被定为二类医疗器械[8]。2018 年2 月至今,定制式AM 医疗器械相关注册技术审查指导原则开始制定。2019年7月4日,中国《定制式医疗器械管理规定(试行)》 正式发布[9]。
目前,国内已发布的AM 医疗器械相关注册技术审查指导原则5 项,正在征求意见的4 项,涵盖了金属、高分子材料的骨科植入物及口腔相关器械。截至2023 年7 月23 日已发布和正在征求意见的AM 医疗器械相关指导原则见表3。

表3 截至2023 年7 月23 日已发布和正在征求意见的AM 医疗器械相关指导原则
4 国内AM 医疗器械技术标准及监管体系建设路径思考
4.1 技术标准建设
基于已有的AM 医疗器械技术标准体系,通过比对AM 工业制造标准,筛选缺乏的医用标准体系。从基础标准、数据/软件、设备、材料、工艺/方法5 个部分全方面比对国内外标准,梳理出工业化标准中适合医疗器械的部分标准,结合现有医疗标准和AM 标准,形成标准体系间的无缝连接,见图4。
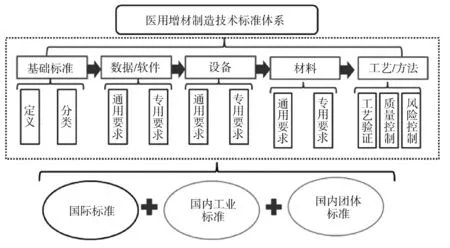
图4 医用AM 技术标准体系
(1)完善医用AM 原材料标准:普通工业AM耗材与医用耗材标准有明显区别,结合已有的医用植入物耗材国家标准、行业标准,根据风险等级对工业化AM 耗材中的性能指标重要性进行分类。(2)完善工艺验证方法标准:从加工工艺、专用力学性能测试方法、化学特性测试方法及生物学特殊风险评价方法4 个角度对工艺验证方法标准进行分析,通过查找国内外文献等方式,提出具有针对性的建议,如加工工艺方面(提出稳定性方法标准和后处理工艺稳定性、有效性标准)、专用力学性能测试方法(提出专用测试方法、非破坏性测试方法等)。(3)完善专用设备标准:提出针对不同AM 成型的工艺要求,建立专用于AM 医疗器械打印设备的通用和专用要求,以及设备参数的验证方法。
4.2 监管体系建设
归纳总结AM 医疗器械全生命周期的质量控制相关标准,提炼安全有效的质量控制要点。从产品设计、原材料控制、医工交互、数据建模、设备参数控制、打印后处理、成品质量控制、上市后使用的闭合生命周期,分析其中的质量控制要点。(1)产品设计的质量控制点包括原材料选择确认、打印模型获取途径确认、AM 设备确认。(2)原材料的质量控制点包括化学性能(粉末的化学组成)、物理性能(颗粒形状及结构、粒度分布、比表面积、颗粒密度等)、工艺性能(松装密度、振实密度、流动性等)。(3)医工交互的质量控制点包括匹配临床与定制需求、三维数据获取与确认。(4)数据建模的质量控制点包括数据传输确认、有限元分析确认。(5)设备参数的质量控制点包括AM 设备运行确认、AM 工艺参数确认。(6)打印后处理的质量控制点包括表面硬度与粗糙度处理、残留粉末或支架去除确认、热应力消除及退火等力学性能补强方式。(7)成品质量的控制点包括物理化学性能评价、力学性能评价、生物相容性评价。(8)上市后使用的质量控制点包括不良事件监测。
5 小结
AM 医疗器械主要以二类和三类中高风险产品为主,且每年获批注册证的数量呈上升趋势。相比之下,国内现行AM 医疗器械标准仅有5 项,难以完全覆盖已注册和即将注册的产品。通过参考对比相关工业AM 标准,结合医疗器械产品特点建立适用的AM 医疗器械标准,以丰富现有的标准体系具有可行性。此外,AM 医疗器械产品相关的注册审查指导原则正在不断发布,全生命周期的质量控制仍是审查和监管的主要要点和方式。
猜你喜欢
数学物理学报(2022年5期)2022-10-09 08:58:18
医疗装备(2020年10期)2020-06-13 01:34:36
电子制作(2019年16期)2019-09-27 09:34:56
电子制作(2019年15期)2019-08-27 01:12:02
质量安全与检验检测(2019年3期)2019-07-31 06:37:00
质量安全与检验检测(2018年6期)2018-12-28 06:23:46
北京航空航天大学学报(2017年4期)2017-11-23 05:48:22
中国工程咨询(2017年12期)2017-01-31 02:56:54
中国洗涤用品工业(2016年2期)2016-02-28 19:03:19
安徽地质(2016年4期)2016-02-27 06:18:21
