
5-羟甲基糠醛路线合成2,5-呋喃二甲酸的研究进展
2024-02-28 07:18许智扬
中国塑料 2024年2期
许智扬,祝 钧
(北京工商大学轻工科学与工程学院,北京 100048)
0 前言
随着人们对可持续发展的关注日益增加,研究人员们对可再生资源进行了广泛的探索。其中,FDCA是一种具有长期、广泛的应用潜力的生物基化学品,被专家认为是替代现有石油化学品的可持续化学品的重要候选物质,在聚酯类塑料、涂料、纤维织物等多个领域具有广阔的应用前景。相较于传统的石油化学品,FDCA 具有较低的碳排放、更好的可降解性和更广泛的资源来源,寻找一种绿色高效的FDCA 制备方法成为了研究的热点之一。
目前,合成FDCA 有5 种路线,分别是己糖二酸路线(见图1)、二甘醇酸路线(见图2)、糠酸路线(见图3)、呋喃路线(见图4)和5-羟甲基糠醛(HMF)路线[1]。其中,己糖二酸路线虽然可以通过脱水环化得到产物但副反应多。二甘醇酸路线简单但原料价格昂贵。糠酸路线虽然原材料成本较低,但产物为混合物,不易分离。呋喃路线的经济性较低,其中,由HMF 路线合成FDCA 是目前研究最多、最广泛的,也被认为是最有希望实现FDCA 规模化生产的路线,具有重要的经济效益。HMF 是一种可再生的生物质原料,具有较高的化学活性和广泛的应用潜力。通过将HMF 经过一系列催化反应转化为FDCA,不仅可以实现对生物质资源的高效利用,还可以减少对石油化学品的依赖,降低碳排放和环境污染。

图1 己糖二酸路线合成FDCAFig.1 Synthesis of FDCA by hexosuccinic acid route

图2 二甘醇酸路线合成FDCAFig.2 Synthesis of FDCA by diethylene glycol acid route

图3 糠酸路线合成FDCAFig.3 Synthesis of FDCA by furoic acid route

图4 呋喃路线合成FDCAFig.4 Synthesis of FDCA by furan route
由HMF 合成FDCA 有2 种路线[2](见图5)。2 种路线的中间产物是不同的,具体选择哪种路线取决于反应的条件和生产的要求。在第一种途径中,醛基优先氧化为羧基以形成5-羟甲基糠酸(HMFCA),随后HMFCA 的羟基被氧化为醛基,产生5-甲酰基糠酸(FFCA),FFCA 被进一步氧化为FDCA。在第二种途径中,HMF 的羟基优先氧化为醛基以形成2,5-呋喃二甲醛(DFF),随后DFF 的醛基依次氧化为羧基以通过FFCA产生FDCA。

图5 通过HMF制备FDCA的2种路线Fig.5 Two routes of preparing FDCA through HMF
本文的目的是对通过HMF 路线制备FDCA 的研究进行综合和总结。目前,通过HMF合成FDCA 所使用的催化方法有很多种,主要有直接氧化法、贵金属催化法、过渡金属催化法、光电催化法、生物酶催化法。本文将介绍各种方法目前的研究进展,并讨论制备过程中的挑战和问题。同时,还将对未来进行展望,以推动FDCA的可持续化学合成和应用。
1 直接氧化法
该方法是指直接使用强氧化性的化合物将HMF转化为FDCA 的过程,使用这些催化剂不但会对实验仪器有一定的腐蚀作用,还会产生较多的副产物和废物,如果处理不当,对环境会造成较为严重的污染,处理它们又反过来进一步增加了生产过程中的成本。本段主要介绍的氧化剂为KMnO4、NaClO、H2O2和叔丁基氢过氧化物(t-BuOOH)。
2007年,Miura 等[3]在室温下使用KMnO4作为氧化剂,在NaOH 的水溶液中制备了FDCA。同时,他们在对比了其他高锰酸金属盐的效果后,发现高锰酸钾的效果最好,FDCA 的产率可以达到89%。2011年,陈天明等[4]发现在n(KMnO4)/n(HMF)=2.4/1,反应时间为10 min,反应温度为25 ℃时,FDCA 的产率最高。
2014年,宋开贺等[5]以纳米金属氧化物CuO 和Co3O4为反应的催化剂,次氯酸钠为氧化剂,在常温常压条件下催化氧化HMF 合成FDCA,最佳反应条件下总收率为98%。
H2O2可作为一种绿色氧化剂被用来氧化HMF,大多数杂多酸催化剂不溶于水,但它们可以溶解在H2O2的混合体系中[6-8]。H2O2的作用下,不溶性催化剂会形成可溶性活性物质,并且当H2O2用完时,催化剂沉淀以便于回收。2016年,Li 等[9]使用八钼酸季铵和十二钨酸季铵作为催化剂,通过H2O2直接将HMF转化为FDCA。当与H2O2偶联时,催化剂对FDCA 的选择性和HMF 的转化表现出高效的性能。该条件下,FDCA 的选择性几乎达到100%,HMF转化率达到99.5%。
此外,Hansen 等[10]在含氮促进剂的存在下使用由CuCl和t-BuOOH 组成的改性氧化体系作为氧化剂,将HMF 催化氧化为FDCA。由于FDCA 几乎不溶于反应介质,所以可以通过简单的过滤来促进产物的有效分离。但产物的饱和可能导致FDCA 沉淀到Cu 催化剂上,或形成FDCA-Cu 盐,产生的抑制作用会降低反应的效率[11-12]。这种方法也会产生较多的废物,因此既不清洁也不经济[13]。
以上,除H2O2属于绿色氧化剂,其他氧化剂使用时都会产生副产物和大量废物,选择性与收率也都较低[14]。使用H2O2作为催化剂,结合适当的促进剂,可以高效地将HMF 氧化为FDCA,并且具有较高的选择性和转化率。这种方法也可以减少对传统强氧化剂的依赖,减少废物的产生,具有环境友好型和经济可行性。
2 多相热催化氧化法
HMF 的多相热催化氧化法是指使用多相催化剂将HMF催化氧化为FDCA的过程[14]。
多相催化剂是指在反应体系中以固体或液固两相形式存在的催化剂。相比于传统的均相催化剂,多相催化剂通常具有较大的表面积和丰富的活性位点,能够提供更多的反应活性中心,从而提高催化活性。其结构可以通过调控催化剂的制备方法和条件来进行优化,进一步提高催化活性。
不同的溶剂体系和不同的催化剂选择也都会影响反应路径[15-20]。关于酸性溶剂体系,使用酸性水溶液对于实验设备的腐蚀程度往往较为严重,且通常会因为产生大量副产物使得产率降低。在碱性的水溶液中,使用贵金属催化剂合成FDCA 可以获得可观的产率,部分产率甚至接近100%;碱的加入还可以让生成的FDCA 以盐的形式溶解,使得催化剂表面脱附;但过量的碱在加酸中和处理时会产生废水,不利于大规模的生产[14]。无碱水溶液通常使用碱性载体来负载贵金属催化剂,不需要加入过量的碱。但生成的酸性产物可能会与碱性载体发生反应,影响催化剂的使用寿命[21]。碱性对于无碱水溶液不是必需的,例如碳纳米管载体对于HMF 的吸附作用也可以促进反应进行。对于DMSO、DMF 等有机溶剂的相关研究也取得了不错的进展,在有机溶剂中通过果糖合成HMF 和HMF 合成FDCA 的两步可以不经过分离HMF 连续进行。所以关于有机溶剂的研究在相关反应中有重要价值。
2.1 贵金属氧化法
贵金属催化剂催化HMF 氧化为FDCA 的研究是目前最广泛的。研究表明,贵金属纳米颗粒表面积大、活性位点丰富,具有优异的催化性能。在促进HMF 的氧化反应时,贵金属表面的活性位点起到了至关重要的作用。通过调节催化剂的晶面结构、表面组成和孔道结构等因素,可以有效地调控催化剂的催化活性和选择性。例如,利用氧空位可以有效活化分子氧,与贵金属纳米颗粒进行协同作用的原理,对催化剂进行预处理增加其中的氧空位,可以提高催化剂的性能。
贵金属催化剂还可以通过氧化还原循环来实现反应的连续进行,提高反应的效率和产物的选择性。通过控制合成条件和添加适当的表面修饰剂,可以进一步提高贵金属催化剂的催化活性和稳定性。此外,还可以利用载体材料、复合材料等方法来改善催化剂的分散性和稳定性,提高催化剂的循环使用性能。
目前常用的贵金属催化剂有Pt 催化剂、Au 催化剂、Ru催化剂和Pd催化剂。Pt催化剂因其具有优异的催化性能和稳定性,在许多重要的催化反应中被广泛应用。常见的Pt 催化剂包括Pt 纳米颗粒、Pt 合金催化剂和Pt 负载型催化剂。其中,Pt 纳米颗粒具有高比表面积和活性,能够提高反应速率和产物选择性。Pt 合金催化剂通过与其他金属形成合金,可以调节催化剂的活性和选择性。Pt负载型催化剂通过将Pt颗粒负载在介孔材料或碳材料上,可以提高催化剂的稳定性和再生性。
Miao等[22]将Pt纳米粒子负载在Ce0.8Bi0.2O2-δ上,有效地催化HMF氧化为FDCA(见图6),在室温下30 min内实现了98%的FDCA 产率,即使催化剂被重复使用了5 次,对FDCA 的选择性也没有太大损失。通过在可还原氧化物中掺杂不同价数的杂原子使其产生新的空位,可以调节载体的氧化还原性能,因此含铋的二氧化铈会存在大量的氧空位,此外铋也可以促进过氧化物中间体的裂解,共同加速了氧还原过程。综合以上特点,我们可以知道Bi 的添加会增强催化剂的抗氧化性,同时又能抑制催化过程中Pt的流失,可以增加催化剂的稳定性,延长催化剂的使用寿命[23]。
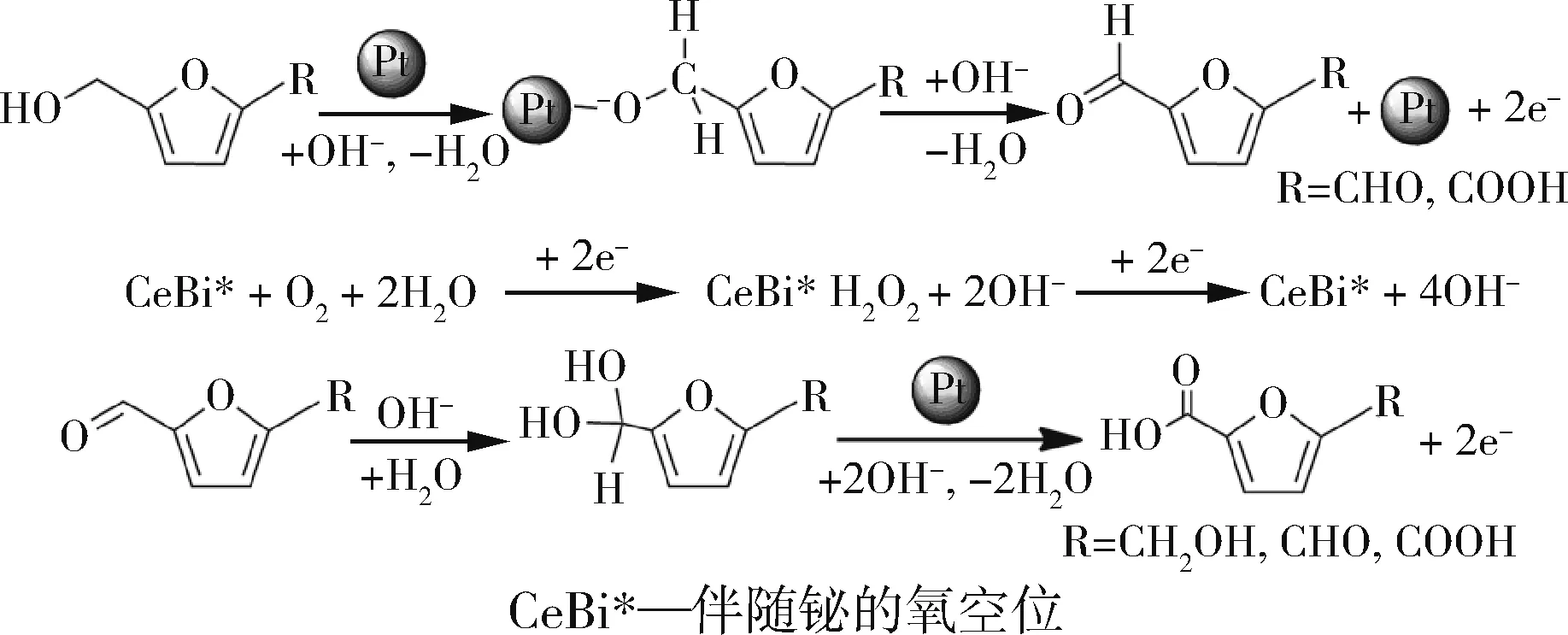
图6 在反应体系中同时存在Pt纳米粒子和Bi掺杂的二氧化铈的情况下HMF氧化的反应机理Fig.6 Reaction mechanism of HMF oxidation in the presence of both Pt nanoparticles and Bi doped cerium dioxide in the reaction system
Zhang 等[24]以Fe3O4为核,和表面由Pt 纳米粒子团簇修饰的保护性无定形碳壳共同组成了具有核壳结构的超顺磁性Pt 纳米粒子催化剂(Fe3O4@C@Pt)并应用于HMF 的氧化。这种催化剂表现出比其他催化剂更高的催化活性,仅在反应4 h 后即可获得接近于100%的FDCA 产率,且合成的微球催化剂可以重复使用3次以上,不会造成显著的性能损失。
Au 催化剂现在被认为是许多有机合成基本反应的活性促进剂,与其他催化剂相比,Au 催化剂突出表现了自身的高选择性。同时,Au 催化剂还表现了其他基本特性,如易于回收性和生物相容性,使用Au 催化剂与直接使用高锰酸盐、次氯酸盐等会产生大量废物的方法相比,显得更加“绿色”。
Taarning 等[25]和Gorbanev 等[26]都使用了Au-TiO2催化氧化HMF 来制备FDCA,前者在加入少量甲醇钠(质量分数为8%)作为碱的条件下以甲醇(1/79 摩尔比)为溶剂,后者在高压氧气和高碱浓度的条件下在水溶液中反应。
除TiO2之外,CeO2也可以作为载体,Casanova等[27]尝试使用CeO2作为载体,他们对Au-CeO2进行了预处理,增加了其中的氧空位和Ce3+的比例,来促进O2的活化和脱氢过程,提高催化剂性能。然后他们将其与活性炭载体金催化剂、Fe2O3载体金催化剂比较在水溶液中催化氧化HMF 所得到FDCA 的产率,在NaOH/HMF 摩尔比等于4,130 ℃,1.0 MPa O2的条件下反应8h 后,所得产率达到99%,突显了CeO2的优势。这证明了与其他负载型纳米颗粒Au 催化剂相比,Au-CeO2是在水中将HMF 好氧氧化成FDCA 的优异催化剂。然而该催化剂需要使用碱且并不稳定,其催化活性在第二次运行中迅速下降。同时他们尝试探究了Au-CeO2的稳定性和可重复利用性,通过在两步升温的过程中进行反应,减少了底物降解并增加了催化剂的寿命。
与Pt、Pd、Au 相比,使用Ru 催化剂的成本要低很多。2017年,Zheng 等[28]使用活性炭作为载体负载Ru纳米颗粒,在Mg(OH)2存在的条件下,383 K 和1.0 MPa O2下FDCA 产率达到97%。在相同的反应条件下,Ru/C 在形成FDCA 方面优于Pt/C 和Pd/C 以及其他负载型Ru 催化剂。碱的存在被证明有助于提高反应速率,但碱并非必不可少。2018年,Pichler等[29]制备了不同的ZrO2负载Ru催化剂,并将其用于在无碱条件下将HMF 氧化为FDCA。使用纯氧作为氧化剂,水作为溶剂,16 h 后实现了HMF 的完全转化,FDCA 产率达到97%。Ru颗粒的尺寸对催化性能至关重要,高表面积ZrO2的利用可以使得Ru颗粒非常小,催化剂因此获得了优异的活性。同年,Ferreira 等[30]使用Ru/Al2O3催化剂,以Na2CO3提供碱性条件(pH=11),在高氧气压力(30 bar O2)下氧化HMF 的FDCA 产率达到了98%,且在每次运行后催化活性完全恢复,从绿色化学的角度来看Ru/Al2O3催化剂有不错的应用前景。
Pd 催化剂具有催化活性高、选择性好、易合成使用、易回收再生、多种反应可行、与其他催化剂配合使用有独特效果、对功能团容忍性好等特点。2015年,Liu等[31]以Fe2O3为核,碳层为壳,组成具有核壳结构的磁性微粒,再将Pd 纳米颗粒固定在上面,制备出催化剂Pd/C@Fe2O3。在优化的反应条件下,HMF 的转化率接近100%,FDCA 产率达到了87.8%。该催化剂的优势在于不需要大量的碱、不需要高氧气压力、完成反应后可以通过磁场进行便捷回收,有利于催化剂的重复使用。
单一贵金属催化剂存在不稳定性等缺点,因此人们引入第二种金属来调整催化剂的结构,进一步增加催化剂的活性和稳定性。Albonetti 等[32]制备出Au-Ce催化剂,将FDCA 的产率提高到了92%。2022年,Cheng等[33]发现Pt-Cu合金纳米颗粒是催化HMF 氧化为FDCA的高活性催化剂,其中Cu到Pt的电子转移增强了催化剂的性能。
贵金属催化剂也存在一些缺点。首先,贵金属催化剂的制备成本较高,这使得贵金属催化剂在大规模工业应用中的成本效益受到限制。其次,在使用过程中贵金属催化剂的寿命会降低,影响催化剂活性和选择性,需要定期对催化剂进行再生或更换。特别是某些对反应条件较为敏感的贵金属催化剂,需要较严格的反应条件才能发挥良好的催化效果。
虽然贵金属催化剂存在一些缺点,但其在有机合成和其他领域的应用仍然非常重要且广泛。随着科学技术的发展,人们正不断努力降低其成本和对环境的影响。
2.2 过渡金属催化剂法
过渡金属催化剂在HMF 氧化制备FDCA 的反应中不但显示出高催化活性、高稳定性,相对于贵金属催化剂来说成本更加低廉。因此其在大规模的工业应用中更具有经济可行性。虽然目前已替代或部分替代贵金属催化剂完成HMF 的选择性氧化,但仍存在一些问题。
Mn 基氧化物具有原材料成本低、结构多样、低毒和环保特性。Hayashi等[34]发现MnO2会与氧气反应生成MnOOH 使反应物活化,使得HMF 完全转化时得到了91%的FDCA 产率。他们还发现碱浓度对产率有影响,随着碱浓度的上升,HMF 的转化率也会变高,但也会生成更多的副产物。Mn 基催化剂的金属氧化物催化体系同样成本低、相较于前者催化效率更高,但是在反应过程中需要使用大量的碱,不但对设备有所损害,还会污染环境。Neatu 等[35]在碱性水溶液中使用Mn/Fe 双金属氧化物作为催化剂,氧化剂为氧气,在Mn 和Fe 物质的量比为3/1 时,FDCA 产率达到90%。在该实验中,碱促进HMF 氧化为FFCA,Mn3+和Mn4+的混合物与赤铁矿相共存,协同作用使FFCA 可以在温和条件下被进一步有效地氧化为FDCA。
铜基催化剂避免了使用碱。Liu 等[36]制备了氨基官能化的SBA-15(SBA-15 是最常见的介孔二氧化硅之一,表面积、孔径和孔体积都较大),并用来固定Cu2+和VO2+,分别得到了SBA-NH2-VO2+和SBA-NH2-Cu2+催化剂,因为氨基官能化的SBA-15对Cu2+等多种金属阳离子表现出较高的亲和力,金属阳离子得以稳定地存在于介孔中。该实验使用氧气作为氧化剂,在110 ℃的甲苯和4-氯甲苯中得到了28.9%的FDCA 产率。最重要的是产物易被分离,催化剂可以循环使用,并且其催化活性没有显著降低。
铁、钴氧化物催化剂不但成本低廉、催化活性较高,其合成也简单,更因为其属于磁性氧化物,也可以通过磁场作用来达到分离产物的目的[37]。但多数相关催化体系都需要高压条件,不易控制。Gawade 等[13]以MnFe2O4为催化剂,过氧化氢叔丁醇(TBHP)为氧化剂,对HMF进行氧化,Mn的价态变化使得催化剂活性较高,在100 ℃下反应5 h所得到的HMF转化率和FDCA 的产率都较高(85%)。该实验在反应中没有使用碱,对环境污染小。且循环使用4次后的活性和选择性几乎没有损失。
3 光电催化氧化法
与传统的化学氧化方法相比,光电催化氧化法通常在室温下进行,无须高温和高压条件;不需要使用传统氧化剂,使用的催化剂通常是可再生、易回收的,对环境友好;可以精确控制反应的速率和产物的选择性,满足不同实际应用的需求。这些优点使得它成为一种有潜力的安全、清洁的绿色催化氧化法。
3.1 电催化法
相较于其他催化方法,电催化法可以通过控制电流强度或电位来调控反应的结果,以便捷地找到最合适的环境与反应条件[38-39]
Cai 等[40]首次研究了镍基二维金属有机骨架(2D MOFs)作为电催化剂将HMF 氧化为FDCA,2D MOFs 是由离子和对苯二甲酸(BDC)配体配位形成的。他们通过溶剂热法成功合成了一系列金属掺杂(Co、Fe、Mn)的2D Ni-MOFs。其中,Co 掺杂的NiCoBDC-NF 表现出优秀的催化活性、选择性和稳定性。FDCA产率为99%,法拉第效率为78.8%。Co2+和配体使得在2D MOFs中容易形成高价态的Ni离子,是醇和醛氧化的活性位点。
Taitt 等[41]系统地比较了NiOOH、CoOOH 和FeOOH 电极在pH=13 的溶液中对HMF 电催化氧化为FDCA 的活性,发现它们具有明显不同的催化能力。结果表明,相较于CoOOH 和FeOOH,NiOOH 是用于电化学氧化HMF 的最有效的催化剂,在1.47 V 相对可逆氢电极下实现了96.0%的FDCA 产率和96.0%的法拉第效率。CoOOH可以在相较于NiOOH 更低的电势下催化HMF 氧化,是因为在低电势下CoOOH 可以发生Co(OH)2/CoOOH 转化,但CoOOH 也只在低电位有优势,这是因为只有在低电位下才会实现该转化;当施加更大的正电势以增加HMF 的氧化速率时,会发生水氧化,从而降低HMF 氧化时的法拉第效率。在低于水氧化起始电位的电位下,FeOOH 对HMF 氧化并不表现出任何催化活性。但在1.71 V恒定电位的条件下进行的HMF 氧化过程中却可以检测到微量FDCA,这表明FeOOH也具有氧化HMF的能力。
催化剂的结构设计对催化活性也有重要作用。Gao 等[42]以导电的NiSe 纳米线为核心,活性NiOx为外壳组成了NiSe@NiOx核壳纳米线,并将其作为氧化HMF 的电催化剂。活性位点为NiOx壳层中的高价态Ni,NiSe 及核壳结构都是可以提高HMF 的转化率的。其表现的Tafel 斜率较小(23 mV/dec),同时还具有99%的法拉第效率,即使在连续电解后仍然显示出强大的稳定性。
Zhang 等[43]制备了NiBx作为催化剂,让对硝基苯酚还原制备对氨基苯酚的同时将HMF 氧化制备FDCA。转化率和FDCA 产率均大于等于99%。这证明了太阳能驱动生物质转化的可行性[15],应该在未来的绿色化学中被具体应用,具有实际意义。
3.2 光催化法
太阳能是取之不尽用之不竭的绿色能源,因此光催化法与其他方法相比蕴含着巨大的潜力。Cha 等[44]研究了一种产氢光电电池PEC,尝试使用太阳能进行生物质转化,阴极为水还原产生氢,在阳极由四甲基哌啶氮氧化物(TEMPO)介导将HMF 氧化为FDCA,获得了大于等于99%的产率和100%的法拉第效率。这表明,太阳能驱动的生物质转化可以是一种可行的阳极反应,有可能提高太阳能电池的效率。
Xu 等[45]报道了一种分散附着在石墨相氮化碳(g-C3N4)上的硫代卟啉钴(CoPz)光催化剂CoPz/g-C3N4,在太阳光下使用空气中的氧分子作为氧化剂,催化HMF 选择性氧化为FDCA(见图7)。在环境温度和空气压力下,在pH=9.18 的水溶液中最高获得了96.1%的FDCA 产率。在pH=4.01 下会生成DFF。因此,可以通过调节反应体系的pH 来选择目标产物。催化剂中CoPz 与g-C3N4之间存在较强的相互作用,可以促进CoPz 在光照下产生1O2(单线态氧1),同时抑制g-C3N4产生羟基自由基的能力(羟基自由基对反应选择性有一定影响),1O2选择性将HMF 氧化为FDCA。此外,该催化剂可以循环使用[46]。

图7 CoPz/g-C3N4光催化氧化HMF制备FDCA的可能机理Fig.7 Possible mechanism of CoPz/g-C3N4 photocatalytic oxidation of HMF to prepare FDCA
Han 等[47]制备了由镍纳米离子修饰的超薄CdS 纳米片催化剂(Ni/CdS)[48-49],见光照射下,HMF 被催化氧化为FDCA,转化率和产率都接近100%。HMF 中醛基与Ni/CdS 的结合亲和力强,选择性好,且催化剂比较稳定。
4 生物催化法
生物催化法是使用酶催化或全细胞催化来促进化学反应的一种方法。其通常在温和的条件下进行,能够以可再生的方式利用废弃物和可再生资源,具有环境友好性和高选择性。这使得生物催化法在多个领域具有广泛的应用前景。
4.1 酶催化法
一种新酶活性的鉴定和开发成功,通常能够开启一个新的生物催化工程[48]。2014年,Dijkman 等[50]发现了一种新的HMF 氧化酶(HMFO)能够高效转化HMF 为FDCA,产率大于95%,虽然在FFCA 阶段经常受挫,但该实验突出了酶在生物催化氧化中的作用。
通常,需要多种酶来氧化HMF 中的羟基和羰基,因为大多数酶都只能做到两者之一[51]。2015年,Carro等[52]报道了真菌芳醇氧化酶(AAO)氧化HMF 的能力,在几个小时内可以将HMF 几乎完全转化为FFCA。之后他们发现AAO 不能直接氧化FFCA 中的羰基,只有少量的FDCA由HMF形成,这可能是AAO产生的H2O2的原因。随后他们通过引入一种真菌非特异性血红素过氧酶(UPO)来完成向FDCA 的转化,通过利用AAO 产生的H2O2,该酶可以将FFCA 转化为FDCA(见图8)。当AAO 生产FFCA 完成时,通过添加UPO,可以得到达91%的FDCA 产率,O2是唯一需要的共底物,水为唯一副产物。
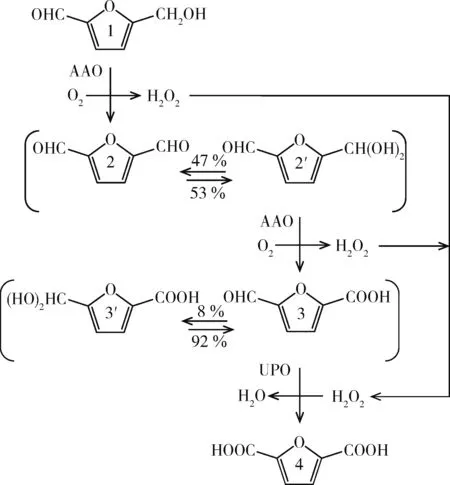
图8 AAO和UPO的作用机理Fig.8 Mechanism of action of AAO and UPO
一锅氧化法具有不需要分离中间产物,效率高的优点。McKanna 等[53]通过控制反应条件,使用半乳糖氧化酶Goase M3-5、周浆醛氧化酶(PaoABC)、过氧化氢酶和辣根过氧化物酶(HRP),通过HMF的一锅反应成功制备了FDCA,产率可高达100%。PaoABC 可以直接氧化FFCA 的醛基为羧基,并且不需要水合过程[54]。但当HMF的浓度过高时,会生成副产物HMFCA。
利用酶与化学结合的方法,能够将酶催化的选择性与化学催化的高效性结合发挥,并且可以达到互补的效果[54]。Wang 等[55]报道了以TEMPO 为介质的磁性漆酶催化剂具有显著的氧化HMF为FDCA 的能力。在最佳反应条件下反应96 h 后,获得了100%的HMF转化率和90.2%的FDCA 产率。磁性漆酶催化剂具有较好的可回收性和稳定性,在6次催化循环后仍保持较好的催化活性(84.8%)。
4.2 全细胞生物催化法
全细胞生物催化法利用完整的微生物体作为生物催化剂,细胞膜提供了一个良好的环境,使得目标产物能够在细胞内稳定转化,耐受性和稳定性高。同时无须对酶进行分离纯化,具有成本低和效率高的优势[48,54]。
Koopman 等[56]从贪铜菌中得到一种新的HMF 氧化还原酶,可将HMF 转化为FDCA。他们将编码该氧化还原酶的hmfH 基因引入恶臭假单胞菌S12 中,得到了全细胞生物催化剂,并将其用作催化HMF 合成FDCA,产率达到97%。
Yang 等[57]分离了线形硬毛藻(Chaetomorpha linum)中能够将HMF 转化为FDCA 的微生物,并将其用作催化HMF 氧化。其中,分离的洋葱伯克霍尔德氏菌H-2 可在28 ℃、初始pH=7 的条件下转化2 000 mg/L 的HMF 为1 276 mg/L 的FDCA。该研究之后,他们在当地校园土壤中富集和分离了能够将HMF 转化为FDCA的辐射耐受甲基杆菌G-2[58],并对其生物转化和生产FDCA 的能力进行了表征。该菌株在26 ℃、初始pH=7 的条件下能将1 000 mg/L 的HMF 完全转化为最大浓度513.9 mg/L 的FDCA。随着反应的进行,FDCA 的量变多,pH 值随之降低。但洋葱伯克霍尔德氏菌H-2 和辐射耐受甲基杆菌G-2 在pH=7 的环境中才具有优良的催化活性,不过增加底物后,pH 值对催化活性的影响会降低,这说明其对HMF 催化氧化的大规模工业应用具有帮助。
Rajesh 等[59]首次报道了黄曲霉生产FDCA。他们从土壤样品中分离微生物,并分别评估其催化活性。在分离物中,黄曲霉APLS-1菌株被发现可以有效促进HMF 向FDCA 的转化,具体表现为在14 d 内将1 g/L的HMF 转化为0.83 g/L 的FDCA。他们还发现高于1 g/L 的HMF 浓度对生物体有毒,并且更高的细胞浓度使氧受限并降低FDCA的产率。
Yang 等[60]在Krystof 等[61]实验的基础上利用睾丸酮丛毛单胞菌(testosteroni Comamonas SC1588)细胞和漆酶-TEMPO系统,将HMF一锅法合成FDCA。该反应通过HMFCA 的路径。HMFCA 的形成使得pH值降低到酸性范围,有利于漆酶-TEMPO 的催化氧化,在36 h 内获得了87%的FDCA 产率。Yuan 等[62]将HMFO 和hmfH 的基因引入解鸟氨酸拉乌尔菌(Raoultella ornithinolytica BF60),通过基因的共同表达来优化生产FDCA。相较于仅使用解鸟氨酸拉乌尔菌,大大增加了HMF的转化率和FDCA的产率。
但全细胞生物催化法仍存在缺陷,氧气在水中的溶解度本身就有限,细胞还会与反应争夺O2,这是一项不可避免的挑战。目前,双酶级联氧化可能会解决这一问题[63],该方法具有较低的O2依赖性,再通过H2O2的内循环,可能会实现O2的最大化有效利用。
5 结语
通过HMF 路线制备FDCA 是一项具有重要意义的研究工作。实现大规模的工业应用仍面临着一些挑战,贵金属催化法虽然催化效率较高,但成本较高,不适用于大规模工业生产;过渡金属催化法较前者成本低廉,但研究不如前者完善,反应条件苛刻,也存在污染环境的情况;电催化法易控制催化剂活性和目标产物,但所需的电解质使后处理增加了成本;光催化法所使用的能源较为环保,但能量转化率较低;生物催化法虽然选择性高且反应条件温和,但反应周期长。综合来看,光电催化法和生物催化法属于新型催化法,更符合可持续发展的理念。
一种可能的改进方法是设计并合成具有特定结构的新型催化剂,以提高催化反应的效率和选择性。在设计新的催化体系时,最好满足成本低廉且催化活性可观,同时应该注意环境友好性,例如尽量减少碱的使用。重点可以研究新型生物催化剂,生物催化剂优势巨大但研究存在瓶颈,新生物催化剂的研究可以实现高效可持续的FDCA合成方法。
另一个改进的方向是改善现有催化剂的稳定性。由于FDCA 的制备过程中可能会发生反应催化剂的失活和寿命的降低,因此需要提高催化剂的稳定性和抗失活性能。可以通过改变催化剂的结构、调控催化反应条件等手段来提高催化剂的稳定性。
为了提高FDCA 的产率和纯度,未来的研究工作还可以集中在优化反应条件方面。一方面,可以通过调节反应温度和反应时间来提高FDCA 的产率和纯度。另一方面,可以研究不同的溶剂和反应气氛对FDCA 合成的影响,并选择最适合的条件以提高FDCA的产率和纯度。
在FDCA 的合成过程中,产生了一系列中间体,这些中间体的分离和纯化直接影响FDCA 的产率和纯度,合适的分离和纯化方法可降低生产成本和环境影响。一种可能的解决方案是应用新型的分离和纯化技术,实现对中间体的高效分离和纯化,并循环利用溶剂和催化剂,减少废物的产生。另外,开发高效的、可以选择性地转化中间体的新催化剂来减少或避免中间体的产生,也可以简化分离和纯化的过程。
总之,随着新催化剂的设计与合成、现有催化剂的改进、反应条件的优化以及中间体的纯化和分离方法的发展,FDCA 制备方法的效率和可持续性将得到进一步提高,推进其大规模产业化进程。
猜你喜欢
分子催化(2022年1期)2022-11-02
贵金属(2021年1期)2021-07-26
贵金属(2021年1期)2021-07-26
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26
云南化工(2020年11期)2021-01-14
应用化工(2020年9期)2020-09-29
淮南师范学院学报(2015年3期)2015-03-22
河北科技大学学报(2015年5期)2015-03-11
中南民族大学学报(自然科学版)(2014年4期)2014-08-06
无机化学学报(2014年4期)2014-02-28
