
基于A-to-I RNA编辑的列线图预测肺腺癌患者的总生存期
2024-02-25 01:06:08朱鑫杰包德荣徐玉芬
浙江临床医学 2024年1期
朱鑫杰 包德荣 徐玉芬
肺癌是世界上病死率较高的恶性肿瘤之一[1]。而肺腺癌(LUAD)占所有肺癌诊断的50%,近年来其比例不断增加[2]。尽管早期检测和个体化治疗已经得到有效发展,但相当一部分患者仍会出现复发和不良临床结局[3]。近年来大量研究应用基因表达建立肿瘤预后预测模型来判断肿瘤患者的预后情况[4-7],由于基因表达的移动性和检测不稳定性,其可靠性仍有待考量。RNA 编辑(RNA editing)是指在转录后水平上发生的核苷酸插入、缺失或替代,导致核苷酸序列发生改变,使其翻译的蛋白质氨基酸序列、结构、功能或表达水平等不同于原基因序列所携带遗传信息的生物学现象。迄今为止,人类已经发了超百万个C-to-U、U-to-C、A-to-Ⅰ等RNA 编辑现象[8-10]。RNA 编辑相较于基因表达更有肿瘤特异性[11],这使得其在预测预后和潜在治疗位点方面有巨大潜力。在此,作者建立了一个应用ATIRE 预测LUAD 患者总生存期(OS)的预测模型。初步探索这些ATIRE 位点影响LUAD 存活的潜在机制。
1 资料与方法
1.1 资料收集 从TCGA 数据库(https://portal.gdc.cancer.gov/)下载LUAD 患者肿瘤组织和正常组织的转录组数据和临床信息。TCGA 肺腺癌数据库有54 个为正常样本,501 个为肿瘤样本。用PERL 软件提取临床信息,包括:样品名称、生存时间、生存状态、年龄、性别、分级、分期。从Synapse 网站(https://www.synapse.org/#!Synapse:syn2374375/files/)下载TCGALUAD 样品的RNA 编辑数据,并采用PERL 软件删除缺失值>30%的数据。删除表达量过低的样本,然后将RNA 编辑数据和生存数据合并。
1.2 预后模型的构建 501 个肿瘤样本中22 个因缺少对应的ATIRE 数据被排除,故仅有479 个样本被纳入本研究,并采用R 软件中的createDataPartition 函数将样本按照6 ∶4 随机分为建模组(n=288)和验证组(n=191)。通过单因素COX 回归分析初筛肺腺癌患者预后相关RNA 编辑,再通过套索算法(least absolute shrinkage and selection operator,Lasso)回归对建模组的肺腺癌患者预后相关RNA 编辑进行降维,并利用多元逐步COX回归模型筛选出最优的RNA编辑构建肺腺癌的预后模型,得到模型的公式,并获得每个样本的风险分数(Risk score)。基于获得Risk score 的中位数,将建模组患者分为高风险组及低风险组。同时将验证组的样品根据Risk score 中位数,划分为高低风险两组。应用建模组的数据对预测模型进行检验,并采用ROC 曲线及校正曲线显示预测模型在建模组和验证组中预测模型的效能。采用Kaplan-Meier 法对建模组、验证组进行生存分析以及对模型RNA 编辑进行生存分析。
1.3 列线图构建 采用单因素及多因素COX 风险回归,将上述获得的Risk score 与患者的临床特征(年龄、性别、临床分期、TNM 分期)进行独立预后分析,151个样本因临床数据缺失而被剔除,最终328 个样本用于获得肺腺癌患者独立预后因子并构建列线图。采用校正曲线、ROC 曲线、决策曲线检验该联合模型的效能及临床实用性。
1.4 相关性及差异分析 分析Risk score与ADAR基因表达的相关性、肿瘤组织和正常组织中选定Atire 位点编辑水平的差异、RNA 编辑与肺腺癌驱动基因(EGFR、ROS1、ALK)的相关性。
1.5 统计学方法 采用R 4.2.1 统计软件。R 语言包有Survival、caret、glmnet、survminer、timeROC、dplyr、tidyr、CMplot、pheatmap、limma、ggpubr、regplot、rms、ggDCA、ggplot2、clusterProfiler、org.Hs.eg.db、enrichplot、circlize、RColorBrewer、ComplexHeatmap、reshape2、tidyverse、ggExtra。差异分析采用Wilcoxon 秩和检验,相关性分析采用Pearson 相关。P<0.05 为差异有统计学意义。
2 结果
2.1 TCGA 数据集中LUAD 患者基线资料 见表1。
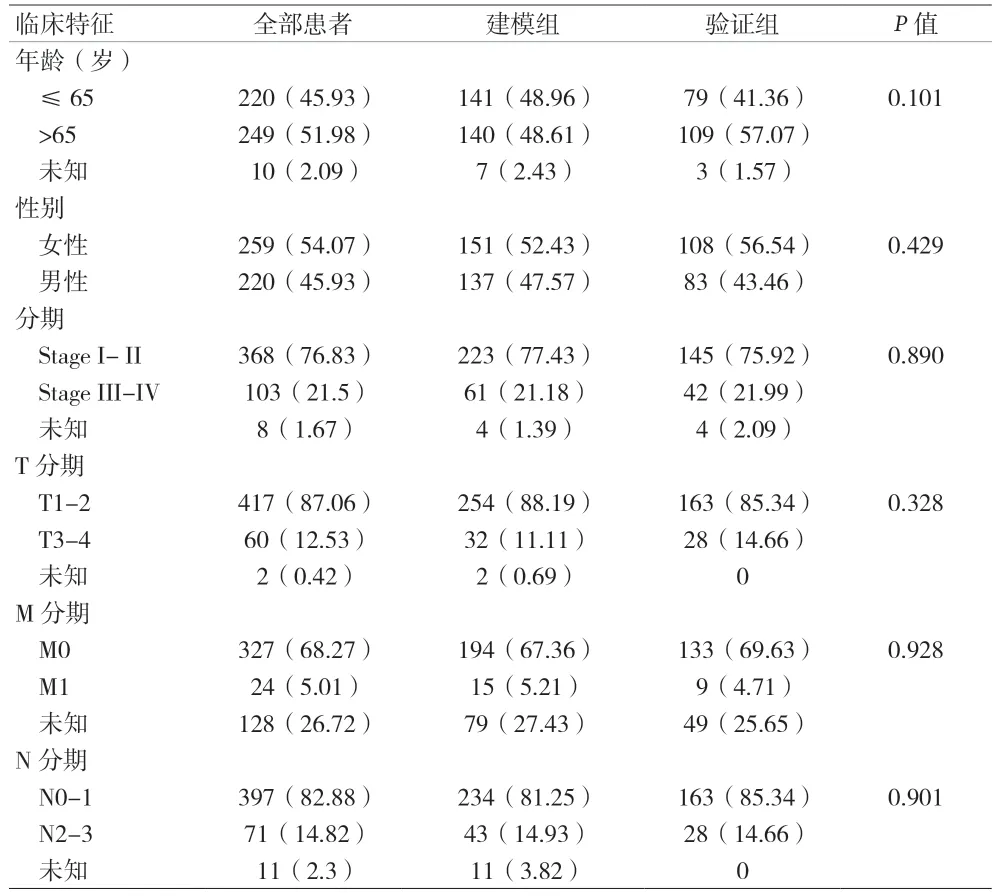
表1 TCGA数据集中LUAD患者临床资料[n(%)]
2.2 预后模型的构建和初步检验 对建模组进行单变量COX 回归分析,共发现8 个ATIRE 位点与LUAD 的OS 强相关(P<0.001),用Lasso 回归分析从中筛选出6个位点,进行多因素COX 分析,剩余4 个位点作为最优预后位点,ADAM19|chr5:156904952、CWF19L1|chr10:101992267、FOXK1|chr7:4809281、CPT1A|chr11:68523468。应用各位点的系数,得到ATIRE 风险评分:(ADAM19|chr5:156904952×8.46)+(CWF19L1|chr10:101992267×1.88)+(FOXK1|chr7:4809281×5.22)+(CPT1A|chr11:68523468×2.96)。4 个ATIRE 位点的风险评分和编辑水平分布在高风险组中明显偏高,随着风险增加生存状态为死亡的患者增加,表明4 个位点可能均与疾病预后不良相关。高风险组的OS 在建模组(P<0.001)、验证组(P<0.001)和所有患者(P=0.038)中均明显降低。模型RNA 编辑的生存分析结果显示,4 个RNA 编辑位点表达量与患者生存时间负相关(P<0.01)。两组中不同年龄、性别、分期、TNM 分期的Risk score差异分析显示风险评分在不同临床特征中差异无统计学意义(P>0.05)。见图1-5。
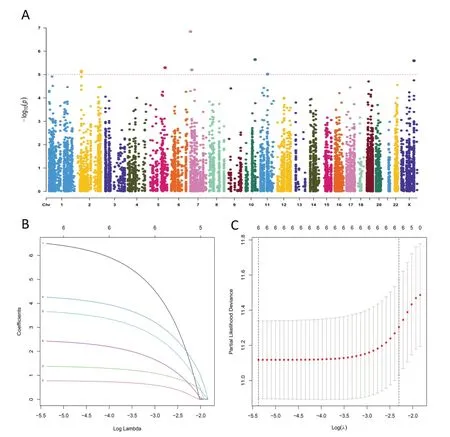
图1 LUAD患者生存相关ATIRE位点的鉴定。A.曼哈顿图描绘了所有ATIRE位点与LUAD生存之间的联系,以单变量COX-PH模型中-log10尺度的P值为x轴,以ATIRE位点的染色体位置为y轴。点橙色线表示P值为0.00001的显著性截断。B.Lasso回归结果;C.选择最佳ATIRE位点(λ)和虚线垂直线的交叉验证
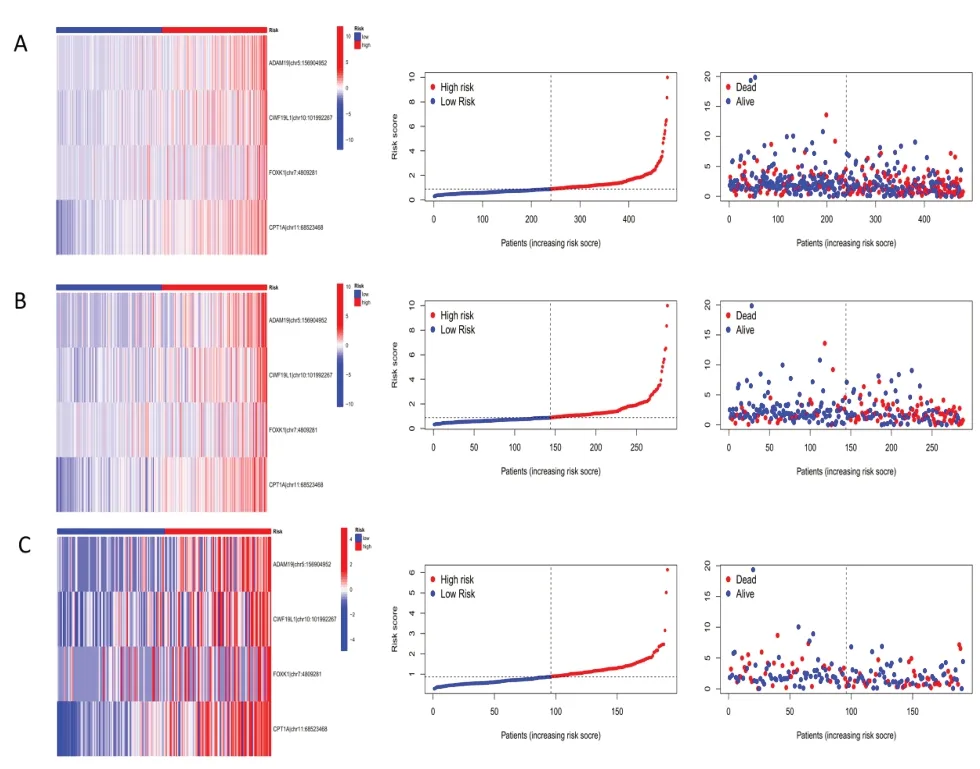
图2 ATIRE风险评分与LUAD患者预后的关系。所有患者(A)、建模组(B)和验证组(C)4个ATIRE位点的ATIRE编辑水平、风险评分、生存状态的分布

图3 所有患者(A)、建模组(B)和验证组(C)中按风险评分分组的生存概率的可视化Kaplan-Meier图
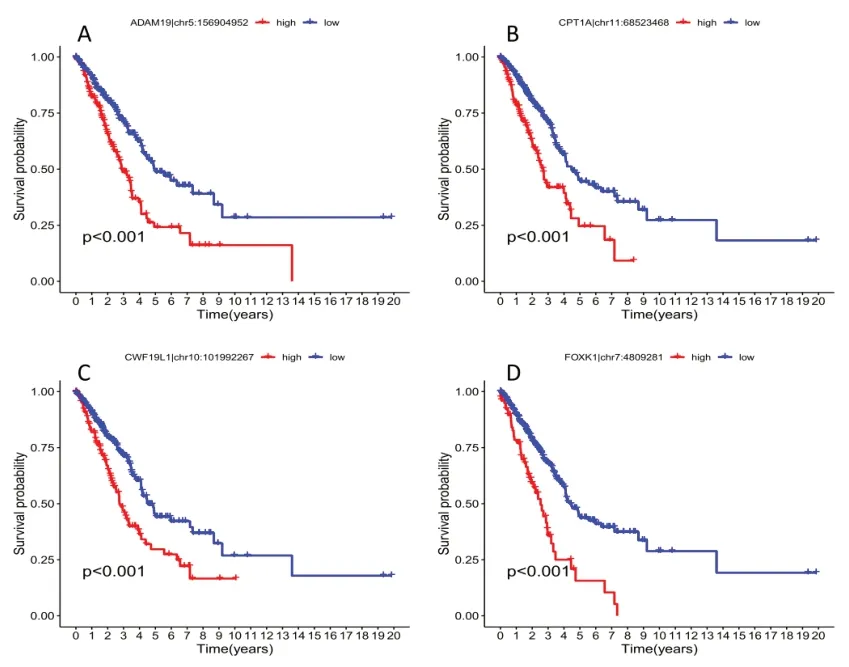
图4 4个ATIRE位点在高低风险组中的生存曲线
2.3 基于ATIRE 的列线图建立及其性能预测 COX 单变量分析结果显示,Risk score 可影响预后。多变量分析显示,Risk score 有成为独立预后因素的潜质。根据ATIRE 风险评分和临床病理特征,包括年龄、性别、临床分期、TNM 分期建立列线图。校准图(95% CI:0.685~0.740)显示在1 年、2 年和3 年观察到OS 与列线图预测的OS 有更好的一致性。ROC 曲线和决策曲线显示Risk(AUC=0.723)和Nomogram(AUC=0.774)大于单一临床病理特征。表明建立的模型比单一临床病理特征具有更高的净效益,在预测患者OS 方面可能存在进一步研究的价值见图6-7。
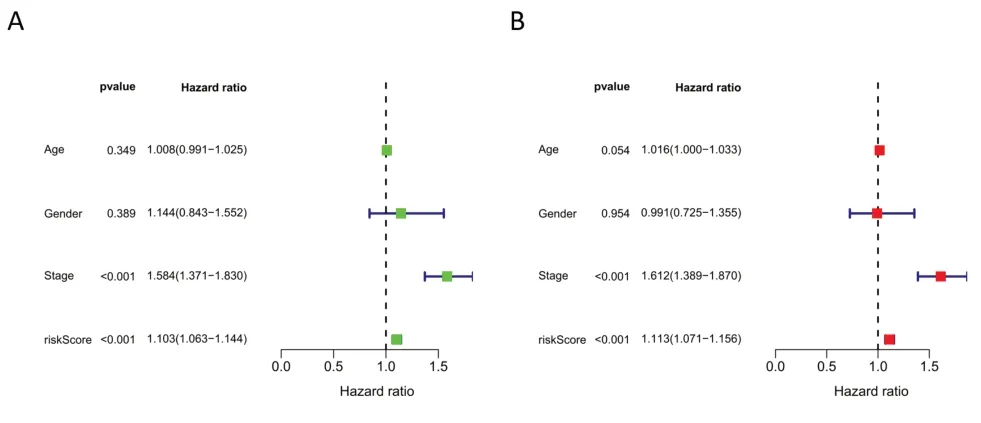
图6 单因素(A)及多因素(B)COX回归分析
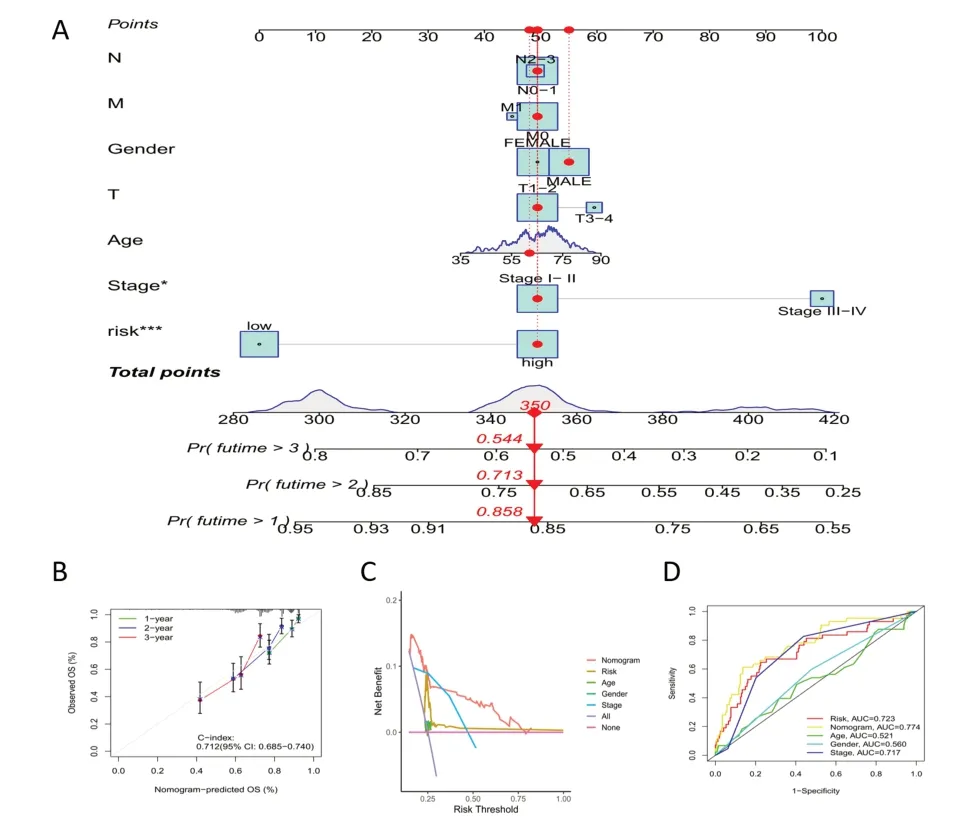
图7 基于ATIRE风险评分和临床病理特征的预后列线图的性能。A.预测LUAD患者1、2、3年OS概率的列线图;B.校正曲线:在1年、2年和3年观察到OS与列线图预测的OS一致;C.决策曲线;D.ROC曲线:不同列线图在预测1年OS的净效益方面的比较
2.4 ATIRE 风险评分和ADAR1 基因表达相关性分析 ATIRE 风险评分和ADAR1 在TCGA-LUAD 肿瘤组织中的表达存在显著正相关(P<0.001)。见图8A。
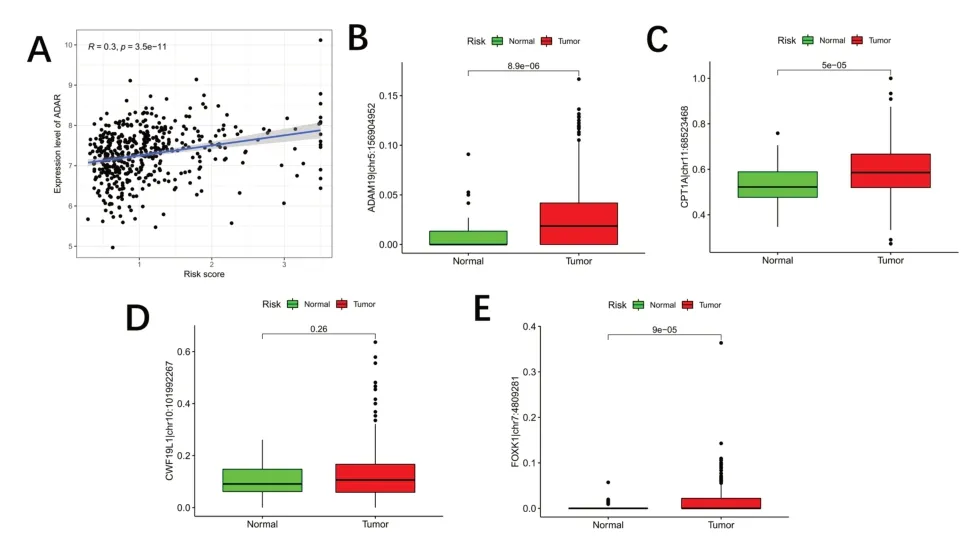
图8 风险评分与ADAR的关系和不同组织间RNA编辑情况的差异。A.风险评分与ADAR基因表达的相关性;B-E.LUAD肿瘤组织和正常组织中选定ATIRE位点编辑水平的差异
2.5 RNA 编辑差异分析 CPT1A|chr11:68523468(P<0.001)、FOXK1|chr7 :4809281(P<0.001)、ADAM19|chr5:156904952(P<0.001)ATIRE 位点编辑水平在肿瘤组织和正常组织间差异有统计学意义。而CWF19L1|chr10:101992267 差异无统计学意义(P=0.26)。见图8B-E。
2.6 RNA 编辑和肺腺癌驱动基因(EGFR、ROS1、ALK)表达相关性分析 RNA 编辑ADAM19|chr5:156904952与ALK 基因表达呈负相关(R=-0.13,P=0.0041)与ROS1 同样呈负相关(R=-0.1,P=0.023)、CPT1A|chr11:68523468 与ROS1 呈负相关(R=-0.13,P=0.0045)、FOXK1|chr7:4809281 与ROS1 表达呈负相关(R=-0.11,P=0.013)、CWF19L1|chr10:101992267 与三个驱动的相关性均不显著。见图9。
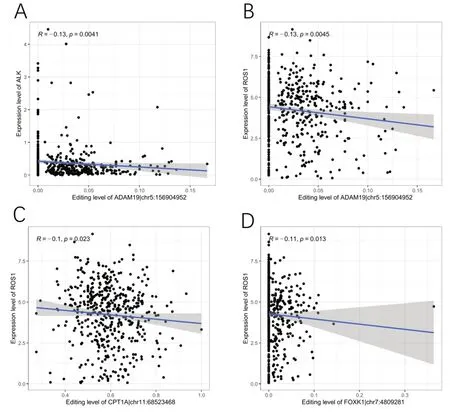
图9 RNA编辑和肺腺癌驱动基因(EGFR、ROS1、ALK)表达相关性 A-B.ADAM19|chr5:156904952与ALK、ROS1表达的关系;C.CPT1A|chr11:68523468与ROS1表达的关系;D.FOXK1|chr7:4809281与ROS1表达的相关性
3 讨论
本研究通过COX-PH 回归和Lasso 算法,确定了4 个与OS 相关的Atire 位点是LUAD 的最佳预后因素。RNA 编辑是一种特殊的转录后修饰,其使得同一个基因翻译出功能相关但又有结构差异的蛋白。其可能与一些功能已知的基因在肿瘤的形成过程中表现出未知的行为有关。如ADAM19 基因编码ADAM(崩解素和金属蛋白酶结构域)家族的成员。其已被证明是一种活性金属蛋白酶,可能参与正常的生理过程,如细胞迁移、细胞黏附、细胞间基质相互作用以及信号转导。既往研究提示正常肺组织也存在RNA 编辑ADAM19|chr5:15690495。ADAM19 能促进部分肿瘤的生长迁移,如其在乳腺浸润性癌(BRCA)中高表达,其上调可促进H2BE76K 细胞的集落形成能力[12]。在结直肠癌中通过siRNA 敲除ADAM19 可以抑制细胞的迁移和侵袭,miR-30c 可以通过直接靶向ADAM19 抑制癌细胞的生长、迁移和侵袭[13]。肺癌中也有关于将miR-153靶向ADAM19 抑制人非小细胞肺癌的迁移和侵袭的报道[14]。相反,在前列腺癌中,ADAM19 在肿瘤细胞中低表达,这显示人类前列腺癌的保护性生物标志物[15]。卵巢癌中ADAM19 的表观遗传抑制可能有助于卵巢癌的进展[16]。
CWF19L1 基因编码CWF19 蛋白家族成员。其可通过替代剪接产生多个转录本变体。该基因的突变与常染色体隐性脊髓小脑性共济失调-17 和轻度认知障碍有关[17-18]。CWF19L1 与人类癌症发生发展的相关性不明确,其致瘤与否及其机制有待进一步研究。
FOXK1 具有DNA 结合转录阻遏活性,RNA 聚合酶II 特异性和转录顺式调控区结合活性。参与多个生物学过程,包括细胞葡萄糖稳态、自噬的负调节、转录的调节和DNA 模板。FOXK1 激活能促进三阴性乳腺癌的淋巴管生成和转移[19]。FOXK1 过表达可以增强细胞迁移能力和侵袭能力[20]。敲低FOXK1 可通过抑制糖酵解抑制肝癌细胞活力[21]。高FOXK1 表达与肝细胞癌不良预后相关[22]。FOXK1 表达与肝癌患者OS 成反比[23]
CPT1A 是肉碱依赖性转运线粒体内膜的关键酶,其缺乏导致脂肪酸β-氧化速率降低。其对多种癌症的影响也是基于影响脂肪酸β-氧化实现。如miR-328-3p—CPT1A—脂肪酸在β-氧化干性轴中负责乳腺癌转移[24]。卵巢癌中较高的脂肪酸氧化途径表达与铂抗性有关,且肉碱棕榈酰转移酶1A 的抑制使卵巢癌细胞对铂敏感[25]。CPT1A 通过调节脂肪酸氧化抑制凋亡促进大肠癌细胞转移[26]。前列腺癌中雄激素减少促进CPT1A 高表达肿瘤的生长[27]。
ATIRE 可能导致非同义的氨基酸突变,选择性剪接的错误调节,密码子偏好紊乱以及microRNA-mRNA重定向或RNA 结合蛋白-mRNA 重定向,从而影响基因原有功能,这导致肿瘤细胞的增殖、侵袭、迁移和耐药能力增强。同时,肿瘤组织中ATIRE 风险评分和ADAR1 显著相关性,提示高风险组中RNA 编辑水平增高。深入研究如何减少肿瘤进展相关的不良编辑事件可能有助于解决肿瘤治疗中的热点问题。
此外,在LUAD 肿瘤组织和正常组织间观察到CPT1A|chr11:6852346、FOXK1|chr7:4809281、ADAM19|chr5:15690495 的编辑水平差异有统计学意义,表明这些异常的编辑可能与LUAD 的发生发展相关。目前认为肺腺癌的驱动基因(EGFR、ALK、ROS1)突变或扩增是预后良好的预测生物标记物,而这些基因的阴性则缺少相应的靶向治疗,这些患者的预后相对较差。RNA 编辑ADAM19|chr5:156904952 与ALK和ROS1 基因表达呈负相关、CPT1A|chr11:68523468与ROS1 呈负相关、FOXK1|chr7:4809281 与ROS1 表达呈负相关可能提示患者瘤内RNA 编辑水平高将影响EGFR、ALK、ROS1 的表达,从而引起不良预后。虽然两者间的相关性仍缺乏相关报道,但其结果也表明模型在预测预后方面具有一定价值。
通常,列线图在预测LUAD 的OS 方面具有中等精度,与预测1 年和3 年OS 的T、N、M 分期系统相比,显示出更好的整体净收益。尽管就有效性而言,与Harrell 的C 指数所示的基于基因表达的列线图相比,ATIRE 的列线图并未显示出明显优势,但就测定可靠性而言,ATIRE 测定相对不易受到RNA 质量和PCR 反应的影响,这可以避免个体间和个体内的变异。
总之,本研究首次建立与LUAD 患者的OS 和临床分期相关的ATIRE 风险评分。虽然结合ATIRE 风险评分和临床病理特征的列线图能用于预测LUAD 的 OS,且在理论上具有较好的效用,但这仍需要大量的真实世界研究来验证该模型的准确性。
猜你喜欢
中学生数理化·八年级数学人教版(2024年8期)2024-08-21 00:00:00
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
供水技术(2020年6期)2020-03-17 08:18:36
生物学通报(2019年3期)2019-02-17 18:03:58
中成药(2018年7期)2018-08-04 06:04:18
东北史地(学问)(2016年6期)2016-12-14 02:03:20
西南军医(2016年3期)2016-01-23 02:17:47
中国当代医药(2015年17期)2015-03-01 02:03:36
西南军医(2014年5期)2014-04-25 07:42:32
