
基于图卷积的离子液体CO2溶解度可解释性预测
2024-02-21 03:47:40张茜茜
计算机技术与发展 2024年2期
张茜茜,陈 平
(1.中北大学 信息与通信工程学院,山西 太原 030051;2.信息探测与处理山西省重点实验室(中北大学),山西 太原 030051)
0 引 言
离子液体是一种理想的绿色CO2吸收剂[1-2],具有化学稳定性好、腐蚀性低、从中分离CO2需要更少的能量、可供选择的种类比较多等优点[3-5]。CO2在离子液体中的溶解度值是重要信息,可以为筛选、设计满足工业需求的离子液体提供重要指导[6]。目前,获取离子液体中CO2溶解度的主要方法包括实验测量和建模。而通过实验测量大量离子液体的CO2溶解度并不现实,实验过程不仅耗时费力且成本高,故借助模型实现准确的预测具有重要意义。基于热力学或动力学方法构建的模型难以处理大量的性质数据,同时预测准确度不够高、通用性差。因此,根据现有的实验数据开发机器学习模型实现准确、快速、高效的离子液体的CO2溶解度预测是十分必要的。
构建预测模型过程中离子液体的表征方式选取尤为重要,当前的模型构建选择的分子表征方式主要包括经验描述符、简单热物理参数、基团信息以及包含多种物理化学性质值、拓扑性质的分子描述符。
经验描述符需要化学背景以及相关先验知识。简单热物理参数多借助现有的数据集,数据集中包含离子液体的偏心因子、临界性质(临界温度与临界压力)以及分子量等。Moosanezhad-Kermani H等采用GMDH分组数据处理方法,提出了一种数学关联形式的白盒模型,模型输入参数为传统的热力学参数[7]。基团信息是指将离子液体手动分解为若干基团的组合,Song Z等使用基团信息表征离子液体,使用人工神经网络(Artificial Neural Network,ANN)和支持向量机(Support Vector Machines,SVM)构建模型预测离子液体中CO2溶解度[8]。这两种表征方式都受限于数据集,对新的离子液体应用困难。Aghaie M等引入了几何、拓扑、信息指数、基于3D矩阵的描述符和量子化学特征等作为输入参数,用决策树(Decision Tree,DT)与随机森林(Random Forest,RF),并与热力学参数模型进行对比,结果证明相比于热力学参数,结构性质与离子液体的CO2溶解度有更强的相关性[9]。然而大量的分子描述符需要借助专业的软件获取,计算成本高。分子指纹作为容易获取的表征方式,常见于药物分子的虚拟筛选模型中,分子指纹是由结构信息编码而成的高维、稀疏向量,这一过程会造成分子的结构信息表征能力受限,且高维的向量在模型训练中易过拟合。
而图卷积神经网络(Graph Convolutional Neural networks,GCN)进行离子液体的结构特征学习可以解决上述分子表征方式存在的问题。GCN可以从分子图中捕获结构特征信息,已经被应用于生物化学领域的活性分析、性能预测等任务中[10],在这些领域的成功应用证明GCN能够有效学习分子图的结构数据,挖掘化合物分子图中的结构信息实现与分子性质的关联。这些任务与离子液体中CO2溶解度预测任务的共性是要实现结构性质与性质的关联,说明通过GCN实现离子液体的预测任务同样是可行的。
该文以离子液体中的CO2溶解度预测为研究目的,提出了基于注意力图卷积神经网络的全流程建模方法,主要贡献概括如下:
(1)在图嵌入模块引入注意力机制,使模型关注原子的非局部特征信息。
(2)将图卷积神经网络与XGBoost有效组合为预测模型,解决了预测模型容易过拟合的问题。
(3)借助注意力池化层与SHAP对预测模型进行解释,能够筛选出对于CO2溶解度预测重要的离子液体结构片段。
1 CO2溶解度预测模型
1.1 总体框架流程
整体流程见图1。首先将离子液体的分子式转化为GCN所需要的图数据,原子的特征向量经过图卷积后生成新的原子特征向量,然后经过池化转换为整个分子图的特征向量,也就是图嵌入网络生成的离子液体的图嵌入向量。

图1 二氧化碳溶解度预测模型构建及应用流程
1.2 图数据获取
要对分子进行表征,通过软件ChemDraw[11](大多数化学结构软件都支持)获取阳离子、阴离子的简化分子线性输入规范(Simplified Molecular Input Line Entry System,SMILES)。SMILES字符串是一种类似语言的描述,可以通过利用化学信息学工具方便地表达分子二维和三维结构特征,从而用于表征分子特征。然后使用Python中的化学信息学工具包RDKit将SMILES字符串转换为所需要的图数据与Morgan分子指纹。RDKit获取的离子液体的SMILES表达式所包含的图数据包含节点特征(如原子的元素类型、价态等)与结构特征(如键的种类、键是否在环中等)。
1.3 图卷积
训练过程旨在学习这些特征来获得分子的整体特征信息,最终表示为一个固定长度的向量[12]。假设离子液体中包含n个原子,包含所有原子的特征向量组成的n×d维特征矩阵X,以及原子之间的n×n维的邻接矩阵A,X与A也就是GCN模型的输入,H(l)表示所有原子在第l层的特征向量矩阵,H(l+1)表示所有原子经过一次卷积操作之后的特征向量矩阵,GCN层与层之间的传播方式如式1所示:
(1)
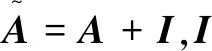
在每一层中,将原子的特征与邻域原子的特征信息聚合,生成新的原子表示,对原子的特征向量进行更新,作为下一层的输入。随着网络层数的增加,逐渐获取所有的原子特征信息与结构信息生成新的原子特征向量。
1.4 注意力池化层
为了实现性质预测任务,希望从离子液体的分子图生成图级表示,大多数先前的研究用简单的最大值池化、平均池化等[14-16]来聚合分子图中所有的原子特征来生成整个分子图的表示,但可能会导致不必要的合并、不完整的解释等问题,因此使用注意力池化层作为传统池化方式的替代。目标是将分子图编码成固定大小的嵌入矩阵,同时最大化原子特征向量所隐含的信息,学习原子的全局信息。
注意力机制允许方法关注神经网络的任务相关部分。对具有序列结构数据的任务应用注意力机制已成为一种惯例,以使模型能够集中注意输入的最相关部分并实现更好的预测[17]。
通过注意力池化层生成注意力系数矩阵,使用注意力机制,将从图卷积模块学习的原子特征向量作为输入来输出权重向量。注意力系数也就是输出的权重,代表不同特征的重要性,权重越大,特征越重要,根据注意力系数大小筛选出对于溶解度预测相对重要的特征,这里的特征是分子图中的化学结构片段,经过图卷积模块的卷积、合并,每个原子的特征向量不仅包含原子本身的信息,还包含原子的邻域信息。
经过图卷积模块,离子液体的结构特征被编码为每个原子的特征向量组合,此时的图表示为G,αvn为原子的特征向量,G的每一行表示一个原子的特征向量,如下式:
G=(αv1,αv2,…,αvn)
(2)
通过注意力池化:
A=softmax(β2tanh(β1GT))
(3)
其中,β1,β2表示权重矩阵,则最终输出的图的表达形式变为:
G'=AG
(4)
加入注意力池化层的图卷积神经网络(Attention Pool Artificial Neural Network,APGCN)所构成的图嵌入生成模块流程如图2所示,通过图卷积神经网络获取分子图中每个原子的特征向量。
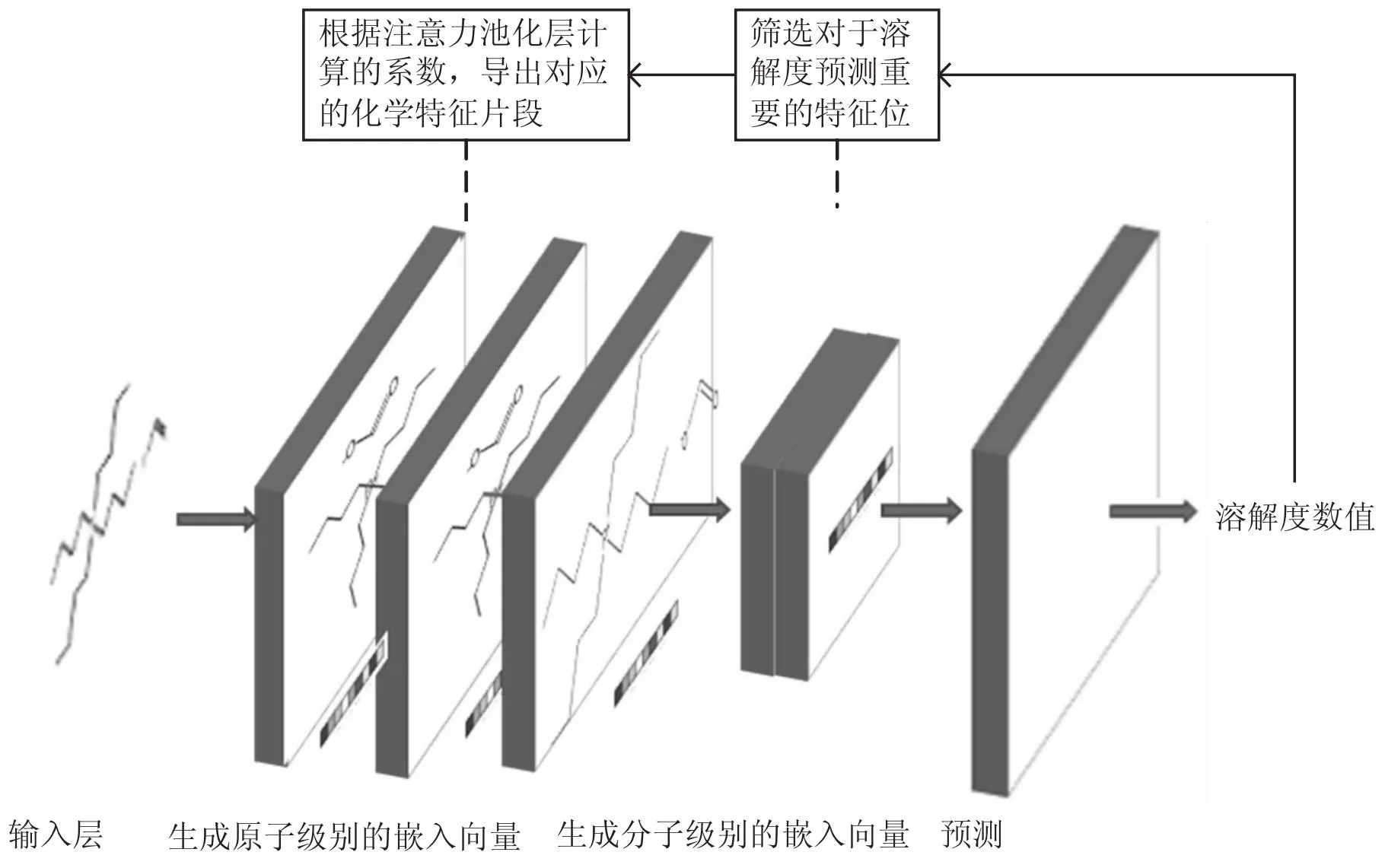
图2 APGCN模块示意图
1.5 预测模块算法选择
目前,离子液体的CO2溶解度预测模型构建中ANN,SVM,RT模型等被广泛应用。在输入数据为分子描述符、高维向量时机器学习算法综合表现更好,训练效率高、不易过拟合[18]。通过6折交叉验证选择最合适的机器学习算法进行建模,在数据集上对机器学习算法XGBoost(e-Xtreme Gradient Boosting)[19]、梯度提升决策树(Gradient Boosting Decision Tree,GBDT)[20]、LightGBM(Light Gradient Boosting Machine)[21]进行6折交叉验证实验对比,评估指标选择决定系数(R2)、平均绝对误差(MAE)、均方误差(MSE)。表1中结果表明,经过验证,在选定的数据集上使用XGBoost构建的模型表现更好,更能避免过拟合,且相比于其他机器学习算法有较高的准确性,同时计算成本低、效率高,故选择XGBoost算法来构建CO2的预测模型。
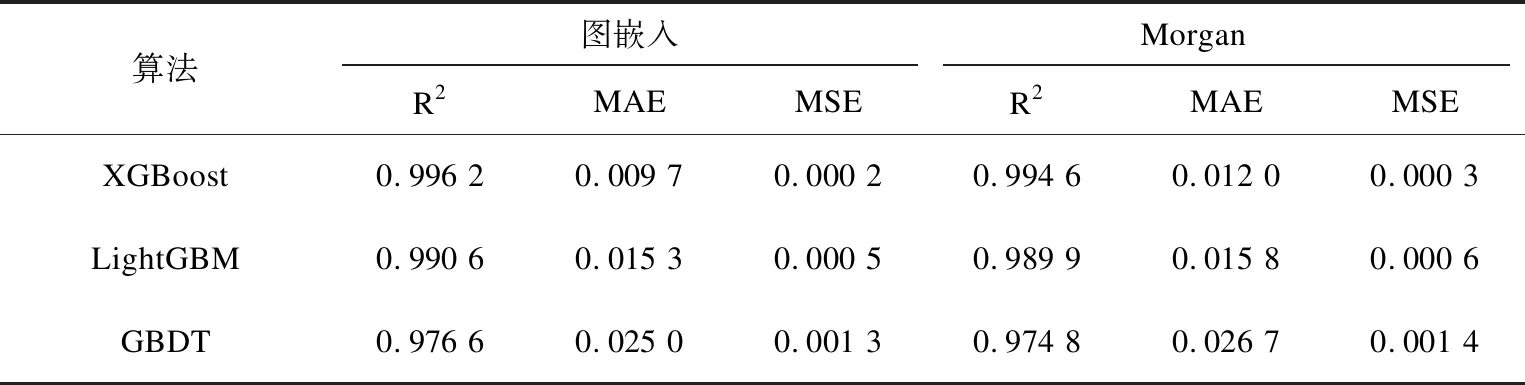
表1 图嵌入和Morgan指纹交叉验证对比
此预测任务的输入不仅有图数据,还有对于离子液体的CO2溶解度不可忽略的重要热力学参数T,P,因此模型要实现基于图数据与数值型混合数据的预测。综上所述,充分考虑到离子液体的CO2预测模型需要输入数据的特点,经过图嵌入生成网络将图数据转化为一个低维向量,将其与对应的T,P条件结合作为XGBoost预测模型的输入,输出最终的CO2溶解度预测值。
XGBoost是基于决策树的集成模型,每次训练时对数据集进行采样,随机抽取一定数量的样本作为每棵树的根节点。每个样本所包含的分子指纹所表示的离子液体结构特征、温度T、压力P即为待分裂的特征。每一棵树生成过程如图3所示,温度T、压力P、结构特征都是影响离子液体CO2溶解度的重要因素,从根节点开始分裂,图3中的树根节点先对压力P进行判断,中间节点有温度T、分子的结构特征向量,对于APGCN-XGBoost模型来说,分子的结构特征向量为APGCN生成的图嵌入向量,对于APGCN-XGBoost模型来说,分子的结构特征向量为Morgan分子指纹,中间节点不断进行分裂生长,直到叶子节点结束。

图3 决策树生成
XGBoost将增益作为特征选择和分裂点选择的准则,增益值越大,说明分裂后能使目标函数减小越多,就越好,用增益值来寻找每次分裂的最优特征和最优分裂点。通过不断地进行特征分裂来生成一棵树,每次添加一棵树,其实是学习一个新函数,去拟合上次预测的残差。在目标函数中加入了正则项控制模型复杂度,以防止过拟合,并对目标函数进行二阶泰勒展开,保留了更多有关目标函数的信息。最终得到XGBoost的最优目标函数如式5所示,
(5)
其中,Gj,Hj分别为损失函数对前t-1轮迭代的一阶导数和二阶导数和,γ和λ为加权因子,ωj为叶子节点权重,T为叶子的个数。
当训练完成后得到了多棵树,对一个样本进行预测时,每棵树的节点对相应的离子液体的分子指纹和温度T、压力P进行判断,最终每棵树都会落到对应的一个叶子节点,模型最终预测的CO2溶解度为每棵树对应的叶子节点之和。
2 实验及结果分析
2.1 实验环境及参数设置
设计APGCN为4层、最终生成的图嵌入维度为50的向量构建APGCN-XGBoost预测模型进行实验。选择半径为8、阳离子和阴离子指纹长度均为512的Morgan分子指纹[22-23]来构建Morgan-XGBoost预测模型进行对比实验,RDKit将SMILES转化Morgan分子指纹,也就是指定长度的二进制比特串。分子结构、T、P对于离子液体的CO2溶解度均为重要的因素,故最终不管是哪种分子表示方式,都需要结合相应的T、P作为预测模型的输入。
超参数优化使用了优化工具Optuna,对基于树的超参数搜索进行了优化,它使用被称为TPE(“Tree-structured Parzen Estimator”)Sampler的方法,这种方法依靠贝叶斯概率来确定哪些超参数选择是最有希望的并迭代调整搜索[24]。反复调用目标函数,找到使目标函数最小化的值。最终优化后模型的超参数见表2。

表2 模型的主要超参数
2.2 实验环境及参数设置
使用的数据来源于Lei等早期的工作[25],其中包含在不同温度和压力下多种离子液体的CO2溶解度数据。共有118种离子液体9 897组CO2溶解度数(0.000 064 8-0.951 6 mol·mol-1),温度243.2-453.15 K,压力0.798 bar-49 990 kPa。阳离子有[BMIM],[EMIM],[MMIM],[HMIM][OMIM],[BMPYR],[N1,4,4,4],[P1,4,4,4]等,阴离子有[BF4][Cl],[DCA],[NO3],[PF6],[SCN],[C(CN)3],[Tf2N],[MeSO4]等。将数据集划分出外部测试集,包含8种离子液体的1 040组溶解度数据,这8种离子液体未被包含在训练集中。将训练集中的实验数据随机划分出20%作为测试集,测试集与外部测试集中的数据均不参与模型的训练。
2.3 评价指标

(6)
(7)
2.4 对比分析
不同模型的相关参数与训练集和测试集的预测性能如表3所示。其中Daryayehsalameh B仅仅对[BMIM][BF4]中CO2溶解度预测建模[26],而APGCN-XGBoost模型可以对多种离子液体实现更准确的预测。
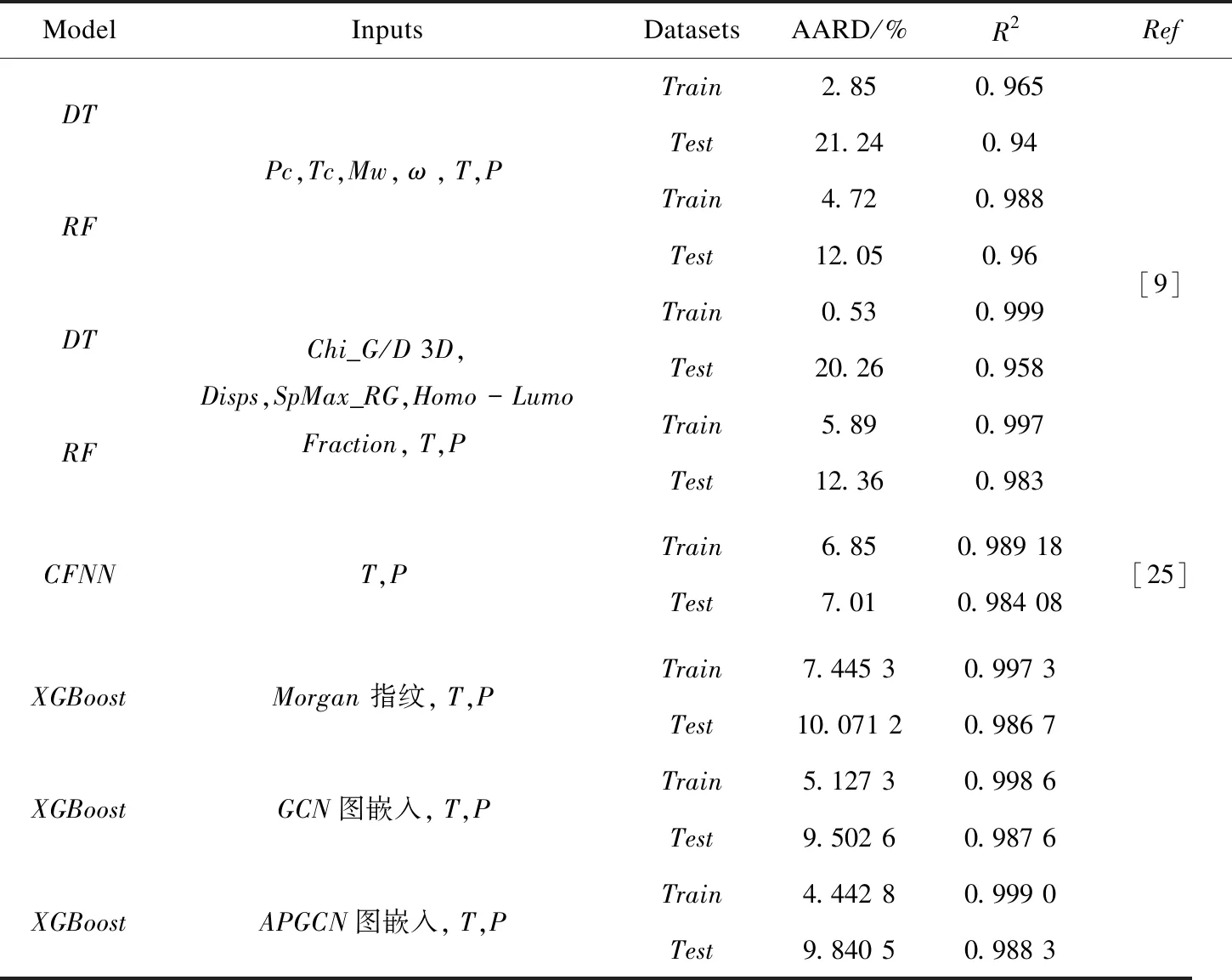
表3 模型的预测结果
由预测结果的对比可以看出,APGCN获取的图嵌入构建的模型测试集的预测结果与实验值的AARD最小,R2最大,能够实现离子液体中CO2溶解度更为准确的预测。APGCN-XGBoost与Morgan-XGBoost,GCN-XGBoost预测模块均采用XGBoost模型,三个模型外部测试集决定系数R2分别为0.943 3,0.729 9,0.924 1,外部测试集的结果说明APGCN-XGBoost模型对于未知的离子液体也能实现更为准确的预测。
APGCN-XGBoost模型不仅在预测准确度方面表现优秀,同时相比于量子化学、分子轨道、拓扑信息、基团信息等描述符的计算,APGCN获取的分子图嵌入无需复杂的计算、专业的软件或者物理化学先验知识,特征工程成本低,易于推广到新的离子液体。综合表明APGCN实现了更好的分子表征,注意力池化层的加入提高了离子液体结构信息的提取效率,提高了预测模型的准确性、泛化性。
训练集与测试集的实验值与APGCN-XGBoost模型预测值进行比较,图4(a)实验值与预测值的拟合效果表明模型能够实现较为准确的预测;图4(b)可以看出测试集大多数误差落在[-0.1,0.1]之间,表明模型的预测值与实验值具有较高的对应性。

图4 实验值和APGCN-XGBoost模型预测的CO2溶解度值比较
实验结果验证了模型的有效性,证明模型可以很好地提取离子液体中的特征信息用于CO2溶解度预测,不仅在准确度上具有明显的优势,且通过图卷积获取的图嵌入维度更低,这能够降低下游的计算量,为预测CO2在离子液体中的溶解度提供了一种便捷、准确的方法。
2.5 模型学习效果
原子的特征向量在学习过程中不断变化,可通过计算不同层中不同原子的特征向量之间的Pearson相关系数(取值为-1到1之间,接近-1或1被称为具有强负相关或正相关),并绘制原子相似性矩阵的热图,以观察学习过程中的变化。以[BBIM][MeSO3]为例,原子的标号为0-17,第0-4层的原子相关性热图见图5,具体标号对应位置如图5(f)所示。在训练之前,如图5(a),不同原子之间相似性混乱,并没有明显的区分,而在训练过程中,具有相似化学特性的原子的特征向量越来越相似,最终整个离子液体按照原子的化学环境分为几个区域,每个区域内的原子的化学特性相似,如图5(e)所示,原子0-3在同一支链上,具有相似的节点特征且随着APGCN层数的增加,几个原子特征向量之间的相关系数增大,最终为1,也就是说原子特征向量之间相似性也随之提高。

图5 [BBIM][MeSO3]的热图
具有相似的节点特征并且在拓扑上彼此接近的原子在最终训练后的嵌入空间中具有更高的相似性。说明APGCN通过学习成功提取了原子化学特性相关的信息,并且能够捕获到原子的非局部效应。
2.6 模型的可解释性
模型不仅能够进行准确的预测,还能对模型的预测机理进行解释,实现结构-性质之间的关联,借助注意力池化层计算的注意力系数与SHAP(SHapley Additive exPlanation)[27]实现模型的解释。首先通过分析温度、压力对模型溶解度预测的影响,验证模型中特征重要性的可信度。SHAP的部分依赖图(partial dependence plot,pdp)可以显示特征与预测值之间的关系。如图6所示,pdp图显示了温度、压力的不同取值与模型预测的溶解度数值之间的关系。可以看出,离子液体中的CO2溶解度随着温度的升高而降低,随着压力的增加而增加,结果与溶解度随着温度和压力变化的理论是一致的,说明模型的预测和解释是可信的。

图6 温度和压力的pdp
用注意力池化层得到的注意力系数来衡量离子液体中结构片段对于溶解度预测的重要性,最终导出对溶解度预测影响较大的结构信息,并将其与已知的结论进行对比。相应的结构信息见图7,分子图中的浅色部分为标注的重要结构片段。从结果可以看出:标注结构均为阴离子,S=O基团与含氟阴离子对溶解度影响较大。已知:离子液体CO2溶解度中阴离子起关键作用[28];S=O基团能够提高离子液体CO2溶解度[29];阴阳离子氟化都能使溶解度升高,阴离子氟化作用更为明显[30]。通过对离子液体CO2溶解度重要结构的可视化,寻找溶解度预测中重要的子结构。将化学常识与模型自动筛选得到的重要特征对比,有利于进一步研究结构与性质的关系,对碳捕集过程中的理想离子液体的筛选与设计具有重要意义。

图7 对模型预测贡献较大的结构片段
从模型的结果可以看出,APGCN能够学习到离子液体分子图中更全局的特征,基于APGCN的离子液体表征方式足够“学习”化学结构与性质之间的关系,并且APGCN-XGBoost模型的开发和使用不依赖于化学反应、化学性质和描述符计算的任何高级先验知识,准确预测的同时还可以指出哪些化学结构与化学特性最相关,可以将这些结构筛选出来,此项工作旨在通过深度学习来解释结构与性质的关联,作为相关离子液体的设计和筛选的辅助工具。这种方法还可以很容易地扩展到许多其他领域,用于多种化学分子的性质预测以及筛选、结构优化工作。
3 结束语
为实现离子液体中CO2溶解度的高效预测,通过图卷积神经网络从离子液体的分子图中捕获有效的特征信息,将APGCN与XGBoost组成高效的预测模型,图数据经过APGCN转化为包含结构特征的低维向量,结合其他参数数据作为输入特征,APGCN-XGBoost模型输出溶解度的预测值。最终结果表明,对于新的离子液体,APGCN能够实现比其他分子表征方式更为准确的预测,且对比于传统的热力学、量子化学、拓扑信息等表征方式特征工程成本低,更容易推广到新的离子液体,说明APGCN实现了更好的分子表征。一方面,APGCN-XGBoost模型能够实现在较宽的温度、压力范围内离子液体CO2溶解度准确的预测,解决了实验测量耗时、成本高、传统模型结构复杂、通用性差、计算成本高等问题,为预测CO2在离子液体中的溶解度提供了一种高效的方法。另一方面,APGCN成功学习到了各个原子和结构的特征信息,这些特征信息不仅可以用于预测离子液体的性质,还可以用于探索不同的原子和结构是如何影响最终的性质预测的,有利于进一步研究结构与性质之间的关系。
猜你喜欢
九江职业技术学院学报(2022年1期)2022-12-02 09:46:54
保定学院学报(2022年2期)2022-04-07 02:26:50
中学生数理化·中考版(2022年12期)2022-02-16 07:37:00
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
许昌学院学报(2018年4期)2018-05-02 12:27:37
中华建设(2017年1期)2017-06-07 02:56:14
中学化学(2016年10期)2017-01-07 08:47:24
中国学术期刊文摘(2016年8期)2016-02-13 13:04:44
