
FTO介导m6A修饰的PRKD2调节SIRT1/HIF-1α通路抑制糖尿病肾病足细胞损伤的机制研究
2024-01-30 01:31李亚宁李成乾青岛大学附属医院内分泌科山东青岛264200
现代检验医学杂志 2024年1期
李亚宁,李成乾(青岛大学附属医院内分泌科,山东青岛 264200)
糖尿病肾病(diabetic kidney disease,DKD)是糖尿病常见的微血管并发症,也是世界终末期肾脏病的第二位原因[1]。目前常见治疗方法如控制血糖或血压,只能实现部分肾保护[2-3]。因此开发新的且有效的治疗方法成为了DKD研究的首要任务。足细胞是终末分化的肾小球上皮细胞,在维持肾小球滤过屏障的完整性方面起着重要作用[4]。研究证实,足细胞功能障碍或损伤是DKD 发生发展的核心事件[5-6]。确定介导足细胞损伤的关键因素将为理解DKD 发病机制提供重要见解。有报道显示,丝氨酸-苏氨酸激酶蛋白激酶D2(serine-threonine kinase protein kinase D2,PRKD2)缺乏会促进胰岛素抵抗和代谢紊乱,引发高胰岛素血症[7]。高胰岛素血症等异常的代谢综合征会导致DKD 发生[8-9]。N6-甲基腺苷(N6-methyladenosine,m6A)甲基化是真核生物mRNA 中普遍存在的修饰,m6A 修饰可以维持mRNA 的稳定性、转运、剪接、定位、翻译和蛋白RNA 相互作用[10-11]。据报道显示,m6A 修饰可以调节炎症和凋亡,是介导DKD 病理损伤的重要机制[6,12]。然而,PRKD2 和m6A 在DKD 发病中的生物学作用尚不清楚。本研究通过高糖诱导小鼠足细胞构建DKD 体外模型,分析了PRKD2 和m6A 去甲基化酶脂肪含量和肥胖相关蛋白(fat mass and obesity-associated protein,FTO)在高糖诱导的小鼠足细胞中的表达及其调控关系,探究FTO 介导的PRKD2 对DKD 进展的调控机制。
1 材料与方法
1.1 研究对象 小鼠足细胞(MPC5)购自中国医学科学院基础医学研究所,在含10 g/dl 胎牛血清和20 U/ml 小鼠重组干扰素-γ(interferon-γ,IFN-γ)的RPMI 1640 培养液中于33℃下培养。
1.2 仪器与试剂 Magna RIP RNA 结合蛋白免疫沉淀试剂盒(17-701,Millipore Sigma);Lipofectamine RNAiMAX试剂盒(13,778-030,Invitrogen Life Technologies);Anti-β-actin 抗体(ab8226,Abcam);Antim6A 抗体(ab284130,Abcam);Anti-FTO(ab280081,Abcam);Anti-SIRT1(ab110304,Abcam);Anti-IgG(ab302644,Abcam);Anti-HIF-1α(ab179483,Abcam);半胱氨酸天冬氨酸特异性蛋白酶3(Cysteinyl aspartate-specific proteinase-3,Caspase-3,G015-1-3),白细胞介素-6(interleukin 6,IL-6,H007-1-2),肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α,H052-1-2)及单核细胞趋化蛋白-1(monocyte chemotactic protein 1,MCP-1,H115) 检测ELISA试剂盒(南京建成);Trizol 试剂(Life Technologies,Carlsbad,CA);ExoQuick exosome 提取试剂盒(System Biosciences,美国);Prime Script RT 反转录试剂盒(Takara,中国大连);miScript SYBR Green 荧光定量PCR 试剂盒(QIAGEN,德国);RIPA 裂解缓冲液和BCA 蛋白测定试剂盒(Beyotime Institute of Biotechnology,上海);ECL 化学发光试剂盒(Thermo Scientific,美国);MK3 型酶标仪(美国Thermo Fisher Scientific 公司);Multizoom AZ100型光学生物显微镜[尼康仪器(上海)有限公司];Centrifuge5804R 型高速冷冻离心机(德国Eppendorf公司);Tanno 5200 型化学发光凝胶成像仪(上海天能科技有限公司);Spectra-Maxi3x型多功能酶标仪(美国MD 公司)。
1.3 方法
1.3.1 细胞培养:未分化的MPC5 细胞在含10 g/dl胎牛血清和20 U/ml 小鼠重组IFN-γ 的RPMI 1640培养液中33 ℃培养。诱导分化时,将足细胞在不含IFN-γ 的非允许条件下37 ℃维持7 天进行实验。建立体外足细胞损伤模型时,将分化的足细胞在含1 g/dl 胎牛血清的培养液中饥饿12h,后接受35 mmol/L 葡萄糖进行高糖刺激24 h。
1.3.2 细胞转染与分组:将高糖诱导后的MPC5 细胞分为control 组、空载体(vector)组及pcDNAPRKD2 组和pcDNA-FTO 组。按照分组,control组加入转染试剂培养;vector 组采用空载体进行转染;pcDNA-PRKD2 组采用PRKD2 过表达载体(pcDNA-PRKD2)进行转染;pcDNA-FTO 组采用FTO 过表达载体(pcDNA-FTO)进行转染;按照Lipofectamine RNAiMAX 试剂盒说明书将上述载体分别转染到MPC5 细胞中。
1.3.3 RT-qPCR 实验:使用Trizol 法提取总RNA,核酸定量后,采用Prime Script RT 试剂盒进行逆转录为cDNA,以此为模版配置PCR 反应体系,使用miScript SYBR Green 荧光定量PCR 试剂盒进行实时分析,条件:95℃ 1min,95℃ 20s,56℃ 10s 和72℃ 15s,32 个循环。引物序列:FTO-F:5’-TCACAG ACGTGGTTTCCGAG-3’,FTO-R:5’-ACCACTGGG TTGAGAGGA GT-3’;PRKD2-F:5’-AGAGCCAGG TAACAGGAACAATAG;PRKD2-R:GTGCTAAGGA GGGAGGCTCT-3’;β-actin-F:5’-CATCCGTAAA GACCTCTATGCCAAC-3’,β-actin-R:5’-ATGGAG CCACCGATCCACA-3’。β-actin 作为内参,通过2-ΔΔCt法计算相对表达水平。
1.3.4 Western blot 分析:用RIPA 裂解缓冲液裂解细胞,通过BCA 蛋白测定试剂盒测量蛋白浓度。将30 μg 蛋白在10g/dl 十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDS-PAGE)上分离,并转移到PVDF 膜上。将膜在室温下用5g/dl 脱脂乳封闭2h,加入Anti-FTO(ab280081,Abcam),Anti-SIRT1(ab110304,Abcam),Anti-HIF-1α(ab179483,Abcam)和Anti-β-actin 抗体(ab8226,Abcam),4℃孵育过夜。次日将膜与辣根过氧化物酶标记的山羊抗兔IgG 在室温下孵育1h。β-actin 作为内参,通过化学发光(ECL)试剂盒对蛋白条带进行可视化,并通过化学发光成像系统进行拍照。
1.3.5 m6A-MeRIP 分析:根据制造商说明,使用Magna RIP RNA 结合蛋白免疫沉淀试剂盒进行RIP。MPC5 细胞在用高糖处理之前用质粒转染。将预先涂有5μg Anti-m6A 抗体或IgG 抗体的磁珠与细胞裂解物在4℃下孵育过夜。然后用蛋白酶K处理含有免疫沉淀的RNA-蛋白质复合物的磁珠以去除蛋白质。使用Trizol 提取RNA,并使用样品进行mRNA 的m6A 免疫纯化,以使用RT-qPCR 进行检测。
1.3.6 ELISA 试剂盒检测IL-6,TNF-α,MCP-1水平和Caspase-3 活性:按照试剂盒使用说明书,分别检测细胞培养液中的IL-6,TNF-α,MCP-1水平以及Caspase-3 活性。
1.3.7 流式细胞术分析:通过膜联蛋白V-FITC/PI细胞凋亡检测试剂盒检测细胞凋亡。将细胞悬浮在膜联蛋白V-FITC 和结合缓冲液的混合物中,室温孵育30 min,然后加入PI 和结合缓冲溶液混匀,室温温育15 min。通过流式细胞仪检测荧光,计算细胞凋亡率。
1.4 统计学分析 采用SPSS 22.0 软件进行统计分析,计量资料以均数±标准差(±s)表示,多组间差异比较采用单因素方差分析(one-way ANOVA),组间两两比较采用LSD 检验;两组间差异比较采用Students-t检验;相关性分析用Pearson 法。P<0.05 为差异具有统计学意义。
2 结果
2.1 高糖诱导的足细胞中FTO 和PRKD2 蛋白表达及相关性研究 Western blots 检测显示,与无高糖诱导对照组相比,高糖诱导的足细胞中FTO 蛋白(0.51±0.04 vs 1.00±0.03)和PRKD2蛋白(0.45±0.03 vs 1.01±0.04)水平显著下调,差异具有统计学意义(t=13.17,16.76,均P<0.001)。Pearson 相关性分析显示,高糖诱导的足细胞中FTO 和PRKD2 蛋白水平呈正相关(r2=0.705 1,P<0.001)。
2.2 FTO过表达转染效率验证 RT-qPCR检测发现,pcDNA-FTO 组FTO mRNA 表达水平(3.21±0.15)较control 组(1.01±0.03)和vector 组(1.03±0.08)显著升高,差异有统计学意义(F=8.35,P<0.001),提示FTO 过表达细胞系构建成功。
2.3 FTO 对m6A 修饰的PRKD2 mRNA 表达的影响 MeRIP 分析结果显示,与vector 组相比,pcDNA-FTO 组PRKD2 mRNA m6A 水平(1.01±0.13 vs 0.56±0.09)显著降低,差异有统计学意义(t=51.37,P<0.001)。RT-qPCR 结果显示,pcDNAFTO 组PRKD2 mRNA 水平(3.16±0.14)较vector组(1.03±0.02)显著升高,差异有统计学意义(t=11.82,P<0.001)。提示过表达FTO 通过抑制PRKD2 mRNA 的m6A 修饰可上调PRKD2 表达。
2.4 RKD2 过表达转染效率验证 RT-qPCR 检测显示,pcDNA-PRKD2 组PRKD2 mRNA 表达水平(3.52±0.21)与control 组(1.03±0.05)和vector组(1.03±0.14)比较显著升高,差异有统计学意义(F=47.13,P<0.001),提示RKD2 过表达细胞系构建成功。
2.5 过表达PRKD2 对足细胞炎症反应及细胞凋亡的影响 见表1。ELISA 法和流式细胞术检测显示,与control 组和vector 组比较,pcDNA-PRKD2 组炎症因子IL-6,TNF-α,趋化因子MCP-1 分泌量、Caspase-3 活性及细胞凋亡率均显著降低,差异具有统计学意义(F=2.35~79.13,均P<0.01)。
表1 过表达PRKD2 对足细胞炎症反应和细胞凋亡的影响(±s)
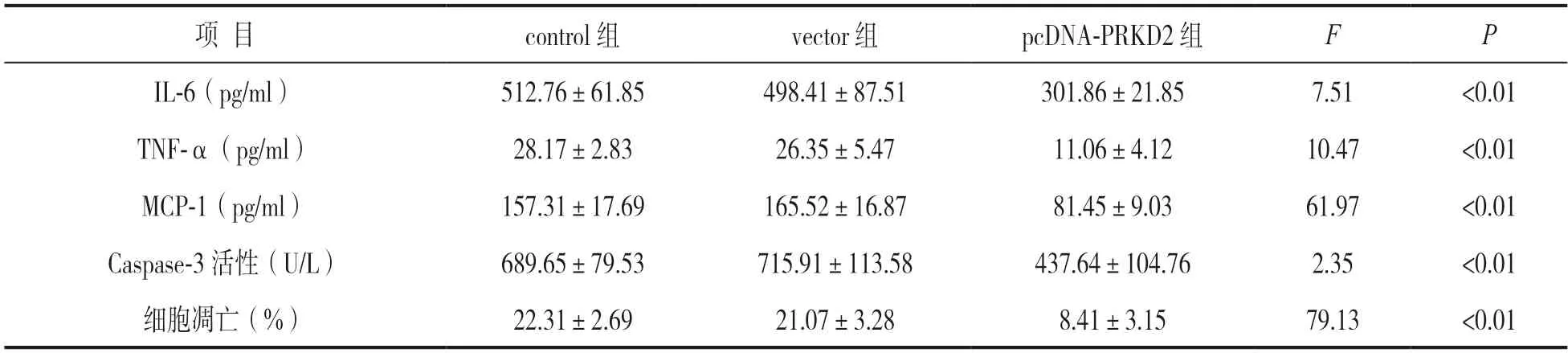
表1 过表达PRKD2 对足细胞炎症反应和细胞凋亡的影响(±s)
项 目control 组vector 组pcDNA-PRKD2 组FP IL-6(pg/ml)512.76±61.85498.41±87.51301.86±21.857.51<0.01 TNF-α(pg/ml)28.17±2.83 7.6926.35±5.4711.06±4.1210.47<0.01 MCP-1(pg/ml)157.31±1165.52±16.8781.45±9.0361.97<0.01 Caspase-3 活性(U/L)689.65±79.53715.91±113.58437.64±104.762.35<0.01细胞凋亡(%)22.31±2.6921.07±3.288.41±3.1579.13<0.01
2.6 过表达PRKD2 对SIRT1/HIF-1α 通路的影响Western blots 检测显示,与control 组(1.01±0.05,1.03±0.07)和vector 组(0.97±0.05,1.02±0.03)比较,pcDNA-PRKD2 组SIRT1 蛋白(3.51±0.15)水平显著升高,HIF-1α 蛋白(0.37±0.07)水平显著降低,差异具有统计学意义(F=31.54,8.31,均P<0.01)。
3 讨论
目前人们普遍认为进行性蛋白尿是DKD 最早的临床特征之一[13]。尿液中清蛋白的存在与足细胞受损和肾小球滤过屏障破坏有关,这种情况称为足细胞病[14]。系膜细胞和足细胞功能障碍会导致糖尿病肾病,足细胞损伤被认为是蛋白尿性肾脏疾病进展的最重要决定因素之一。与许多肾脏疾病一样,DKD 的特征是出现蛋白尿,这是由足细胞凋亡和功能丧失诱导的,随后是与肾小球硬化相关的肾小球滤过率降低[6]。足细胞在支持肾小球结构和功能以及形成滤过屏障方面发挥着重要作用,当足细胞受到葡萄糖或活性氧(read only storoge,ROS)等刺激时,会导致足细胞病中蛋白尿的发生[15-16]。葡萄糖通过诱导E-钙黏蛋白(E-cadherin)和β-钙黏蛋白(β-cadherin)减少,间充质神经型钙黏附蛋白(N-cadherin)的表达及肌成纤维细胞的形成,进而诱导足细胞损伤[15-16]。因此,足细胞耗竭已被作为建立蛋白尿新疗法的新策略。
高血糖是糖尿病并发症的主要因素,高糖诱导的肾脏缺氧是DKD 的常见途径[6]。在1 型或2型糖尿病早期阶段,长期强化血糖控制被认为是糖尿病并发症的强有力的预防策略,尤其是对于DKD。然而,尽管强化血糖控制,约三分之一的糖尿病患者仍进展为DKD,这提出了“代谢记忆”现象。在负责“代谢记忆”的各种机制中,表观遗传修饰(主要包括甲基化修饰、组蛋白修饰和非编码RNA 的表达)引起了更多关注。基于对代谢记忆现象的认识,m6A 甲基化修饰与表观遗传变化的相互作用可导致信号通路持续激活,从而促进DKD 肾脏炎症、纤维化和肾小球肥大的发生。高血糖会增加缺氧诱导因子-1α(hypoxia-inducible factor-1α,HIF-1α)的积累并破坏HIF-1α 的稳定性,通过多种机制导致足细胞损伤[17]。例如,Kang 等报道[18],葡萄糖负载足细胞和糖尿病肾脏中HIF-1α 被诱导,高血糖引起的缺氧通过加速糖尿病足细胞的上皮间质转换和肌成纤维细胞形成导致足细胞损伤。基于以上报道,本研究通过高糖诱导足细胞构建DKD 体外模型,探究m6A 去甲基化酶FTO 介导的PRKD2 对DKD 进展的调控机制。
m6A 修饰是真核生物mRNA 中最丰富的修饰[10]。m6A 修饰的作用包括维持mRNA 稳定性、运输、剪接、定位、翻译和蛋白质-RNA 相互作用。哺乳动物细胞中,m6A 修饰是动态且可逆的,m6A甲基转移酶执行修饰反应,而m6A 去甲基酶则逆转该修饰。特定的RNA 结合蛋白可以直接或间接结合m6A 基序,从而影响RNA 功能。m6A 调节剂的生物学功能与组织发育、细胞分化、昼夜节律和其他多种人类疾病相关,例如癌症进展、糖尿病、神经发育障碍和缺血性心脏病[7,11,19]。此外,m6A修饰可以调节炎症和细胞凋亡,这是介导DKD病理损伤的重要机制。LU 等人[20]的研究发现糖尿病肾病的小鼠肾脏中m6A RNA 水平升高,其甲基转移酶14(methyltransferase 14,METTL14)表达上调;METTL14 通过促进m6A 修饰降解SIRT1 mRNA 来加重足细胞损伤,METTL14 敲低通过促进自噬,减轻体内和体外炎性细胞凋亡,对受损的足细胞产生保护作用。FTO 作为RNA 去甲基化酶,控制m6A 修饰以影响mRNA 的生物合成、衰变和翻译[10,21]。大量研究已证实,FTO 调控miRNA 参与糖尿病并发症的疾病进展,如SUN 等[22]研究发现,过表达FTO 导致m6A 水平降低,通过增加细胞因子信号转导抑制因子1(suppressor of cytokine signaling 1,SOCS1) 蛋白表达,减轻炎症反应和肾损伤,抑制DKD 进展。HU 等[23]研究显示,FTO 介导的m6A 水平降低诱导LncRNA EST00000436340 表达上调,增强了ENST00000436340 与多嘧啶束结合蛋白1(polypyrimidine tract binding protein 1,PTBP1)的结合,进一步导致细胞骨架重排,造成足细胞损伤和DKD 进展。与已有报道相一致,本研究发现过表达FTO 通过介导PRKD2 mRNA 的m6A 修饰可抑制高糖诱导的足细胞炎症反应和细胞凋亡,抑制DKD 进展。
PRKD 家族属于钙模块依赖蛋白激酶超级家族,早期研究表明PRKD 亚型在胰腺的各种外分泌和内分泌细胞中具有不同的表达。现有报道显示,PRKD2通过调节糖代谢参与各种疾病的发病过程,PKD2 是促进膳食脂肪吸收的关键信号节点,其缺失、失活或抑制可改善高脂饮食诱导的肥胖和糖尿病[24]。XIAO 等[25]研究发现,PRKD2 缺乏会增加胰岛素分泌,诱导肥胖和胰岛素抵抗,导致代谢絮乱,引发高胰岛素血症。JIAO 等[7]报道显示,PRKD2 在高脂饮食喂养的小鼠骨骼肌和棕榈酸诱导的C2C12 细胞中表达下调。糖尿病、高胰岛素血症等异常的代谢综合征会导致DKD 发生[8-9]。基于以上报道猜测,PRKD2 可能是抑制DKD 进展的调节因子。本研究对此猜测进行了验证,发现过表达PRKD2 能够抑制高糖诱导的足细胞的炎症反应和细胞凋亡,与早期报道相一致,证实PRKD2 对DKD 进展具有抑制作用。除此之外,JIAO 等[7]报道还显示,PRKD2 在棕榈酸诱导的C2C12 细胞中的表达受m6A 甲基化水平调控,这与本研究结果一致,FTO 通过降低m6A 修饰上调PRKD2 的表达。除此之外,沉默信号调节因子1(silent information regulator 1,SIRT1)/HIF-1α 通路被报道在足细胞相关疾病进展中扮演重要角色[26],如LU 等[20]研究发现,在足细胞病中SIRT1 mRNA 显著降低。CHANG 等[27]研究显示,与非糖尿病小鼠相比,糖尿病小鼠的足细胞中SIRT1 表达减少,调控SIRT1/HIF-1α通路能够改善糖尿病小鼠肾脏足细胞损伤,减轻DKD。SUN 等[28]研究显示,连接蛋白43(connexin 43,Cx43)通过调节SIRT1/HIF-1α 信号通路,抑制高血糖诱导的肾上皮间质转化和肾小管间质纤维化。本研究发现,过表达PRKD2 可促进SIRT1 蛋白表达,降低HIF-1α 蛋白表达,说明PRKD2 调控SIRT1/HIF-1α 通路抑制高糖诱导的足细胞炎症反应和细胞凋亡。但本研究还存在一定的局限性,首先只做了体外实验,FTO 和PRKD2 在体内的调控机制还有待进一步验证;其次PRKD2是否还通过其他途径来调控DKD 的发生还需进行深入研究,以期为DKD 的治疗提供更有价值的治疗靶点,以及更可靠的实验依据。
综上所述,过表达FTO 通过抑制PRKD2 mRNA的m6A 修饰上调PRKD2 蛋白表达,通过SIRT1/HIF-1α 通路抑制高糖诱导的足细胞炎症反应和细胞凋亡。
猜你喜欢
数学物理学报(2021年4期)2021-08-30
新世纪智能(数学备考)(2020年10期)2021-01-04
中成药(2018年6期)2018-07-11
中成药(2017年12期)2018-01-19
中成药(2017年8期)2017-11-22
中国交通信息化(2017年8期)2017-06-06
法医学杂志(2015年4期)2016-01-06
法医学杂志(2015年4期)2016-01-06
长江蔬菜(2015年3期)2015-03-11
中国药理学通报(2014年2期)2014-05-09
