
茶皂素及槐糖脂对紫草素脂质纳米粒子形成和稳定性的影响
2024-01-30 02:11彭盛峰张军兵
食品工业科技 2024年3期
何 洁,彭盛峰,张军兵 ,刘 伟
(1.南昌大学食品学院,江西南昌 330047;2.江西丹霞生物科技股份有限公司,江西鹰潭 335000)
紫草素(Shikonin)是从紫草根中提取出来的天然紫红色疏水性色素,同时也是紫草这种传统中草药中的主要活性成分[1],具有多种生理和药理活性,包括伤口愈合、抗炎、抗菌、抗癌、抗病毒[2-4]等。此外,紫草素还常用在食品[5]、化妆品[6]和纺织品[7]中。在食品工业中作为天然着色剂使用,最大添加量为0.1 g/kg。由于其对pH 具有较好的灵敏性,在酸性条件下呈红色,在碱性条件下呈蓝色,近年来,已成为pH 响应型食品包装显色指示剂[8-10]。然而,由于紫草素自身不稳定,限制了其应用场景与范围。从结构上来看,紫草素的分子结构式为5,8-二羟基-2-((1R)-1-羟基-4-甲基-3-戊烯基)-1,4-萘醌[11],易发生化学反应,对光、热和碱敏感,这是造成紫草素不稳定的主要原因。另一个问题是紫草素是疏水性的,水溶性很低。这两个问题是目前阻碍紫草素这类多酚应用的主要因素[12]。
食品胶体递送系统包括脂质体[13-14]、乳液[15]、纳米粒子[16-18]、胶束[19]和纳米复合物[20-22]等,是解决亲脂活性物质水溶度低和稳定性差最有效的方法之一,且已用于多种生物活性化合物的包封和保护。纳米脂质体由于粒径达到纳米级别,因此具有纳米粒子的效应,也被称为脂质纳米粒子,具有生物利用度高、稳定性高等优点[23],但由于磷脂稳定性差,脂质体膜易受外界影响而被破坏[24]。表面活性剂能够促进胶体粒子的形成,并对胶体粒子的稳定性具有一定作用,这是因为表面活性剂会包裹在粒子周围,在粒子之间产生强烈的静电和空间斥力[25]。根据之前的研究,发现使用茶皂素和槐糖脂包埋姜黄素[26-27]和紫草素[28],提高了其稳定性和生物利用率。在现代食品加工中,人们往往更倾向于使用天然原料。因此,本文选取两种天然表面活性剂茶皂素和槐糖脂,考察对其紫草素脂质纳米粒子的形成、稳定性的影响。近年来,由pH 驱动法制备的多酚纳米颗粒(主要是姜黄素),因其负载能力高、稳定性好和生物利用度高[29]而备受关注。同时,pH 驱动法操作简单、快速、不使用有机试剂,是一种有潜力的食品包埋技术。当pH 大于多酚酚羟基基团电解离常数时,酚羟基去质子化,多酚分子带电量上升,使得其溶于碱液。当多酚碱液与脂质体溶液共混并中和后,酚羟基质子化失去电荷而重新变为疏水,在疏水作用力驱动下,多酚分子进入脂质体囊泡中,实现多酚脂质体的负载[13]。紫草素是一种多酚类物质,与姜黄素相似带有两个酚羟基,紫草素在疏水作用下进入脂质体的疏水膜层中,因此可以采用此技术实现主动包埋[28]。
综上所述,本研究的主要目标是通过pH 驱动法制备负载紫草素的脂质纳米粒子,探讨茶皂素和槐糖脂对紫草素脂质纳米粒子形成和稳定性的影响,提高紫草素水溶解度、稳定性和生物利用率,为紫草素在食品等领域的应用拓展提供理论依据和指导。
1 材料与方法
1.1 材料与仪器
紫草素粉末 95%,陕西文迪生物科技有限公司;卵磷脂 S75,上海阿拉丁生化科技股份有限公司;茶皂素 皂素20%~35%,上海阿拉丁生化科技股份有限公司;槐糖脂 西安博联特化工有限公司;乙醇、磷酸氢二钠、氢氧化钠等 任何其他化学试剂均为分析纯。
M-110P 高压微射流 美国Microfluidics 公司;U-T1810 紫外可见分光光度计 上海屹谱仪器制造有限公司;Zetasizer Nano ZSP 动态光散射电泳仪英国马尔文公司;FD-1-50 冻干机 北京博医康实验仪器有限公司;NANOSURF/C300 原子力电镜 美国BD 公司;D8 AdvanceX 射线衍射仪 德国布鲁克公司;Nicolet IS10 傅立叶红外光谱仪 美国赛默飞公司。
1.2 实验方法
1.2.1 紫草素脂质纳米粒子的制备
1.2.1.1 制备方法 紫草素脂质纳米粒子是由卵磷脂添加天然表面活性剂高压均质混合后,采用pH 驱动法包埋紫草素而制成。参考之前的方法并加以改进[13,26-27],首先将卵磷脂与不同质量天然表面活性剂(茶皂素或槐糖脂)混合搅拌均匀分散于磷酸盐缓冲液(pH6.5,5 mmol/L)中,使溶液中卵磷脂质量分数为1%,茶皂素(或槐糖脂)质量分数分别为0%、0.1%、0.2%、0.5%和1%。待分散完全形成均一体系后,将溶液通过微射流运行三次,运行压力为120 MPa,得到天然表面活性剂修饰的空白脂质纳米粒子。然后将紫草素粉末完全溶解于0.1 mol/L 氢氧化钠溶液,按照所需比例添加到空白脂质纳米粒子溶液中,将混合溶液pH 迅速调至6.5,最终混合液中紫草素浓度为0.1、0.2、0.4、0.6、0.8 mg/mL,置于室温下搅拌平衡0.5 h 后,于8000 r/min 离心30 min 去除未包埋的紫草素颗粒,得到不添加天然表面活性剂的紫草素脂质纳米粒子(Shikonin lipid nanoparticles,SLNP);茶皂素修饰的紫草素脂质纳米粒子(Saponin modified shikonin lipid nanoparticles,Sap-SLNP);槐糖脂修饰的紫草素脂质纳米粒子(Sophorolipid modified shikonin lipid nanoparticles,Sop-SLNP)。将相同质量紫草素粉末直接溶于磷酸盐缓冲液的游离紫草素悬浮液设置为对照组。
1.2.1.2 包封率与负载率测定 使用紫外可见分光光度计在516 nm 处测量紫草素吸光度A,绘制紫草素标准曲线为A=0.0223C-0.001(R²=0.9999),C 为紫草素质量浓度,表明紫草素样品在5~40 μg/mL 浓度范围内具有良好的线性关系。将制备好的样品通过8000 r/min 离心10 min 去除未包埋的紫草素晶体,取上清液用乙醇稀释5~40 倍测量紫草素吸光度,对应包埋的紫草素质量浓度。计算包封率与负载率公式如下:
式中:C1表示离心后上清液紫草素吸光度对应的浓度,mg/mL;C0表示制备样品添加的紫草素吸光度对应的浓度,mg/mL;M1表示离心后上清液中紫草素的质量,mg;M0表示制备时添加紫草素的质量,mg;M 表示卵磷脂与天然表面活性剂总质量,mg。
1.2.2 紫草素脂质纳米粒子的结构表征
1.2.2.1 粒径与电位测定 用磷酸盐缓冲液(pH6.5,5 mmol/L)将样品稀释10 倍后使用动态光散射电泳仪在25 ℃下测定紫草素脂质纳米粒子的平均粒径、粒径分布和Zeta 电位。最终结果以三个样本的平均值和标准差表示,每个样本测量三次。
1.2.2.2 原子力电子显微镜(AFM)分析 用超纯水将样品稀释800 倍后取10 μL 滴在新剥开的硅片表面,自然风干。设置硅悬臂在攻丝模式的力常数为0.58 N·m-1,拍摄样品的微观结构[30]。
1.2.2.3 X 射线衍射(XRD)分析 测量前将样品在10 Pa,-60 ℃条件下冷冻干燥48 h 后研磨成粉末状,设置发散狭缝为1°,入射光束的接收狭缝为0.1 mm。在5°~90°的2θ角范围内进行扫描,扫描速度为2°/min[31]。
1.2.2.4 傅立叶变换红外光谱(FTIR)分析 将样品在10 Pa,-60 ℃条件下冷冻干燥48 h 后按质量比1:100 加入KBr 粉末研磨并挤压成片,设置光谱扫描波数范围为4000~500 cm-1,分辨率为4 cm-1[32]。
1.2.3 紫草素脂质纳米粒子的稳定性研究
1.2.3.1 热和光稳定性 将样品分装于EP 管中,将一批样品置于80 ℃水浴锅中分别加热0、10、20、30、40、50、60 min 后置于冰水中迅速冷却至室温,另一批样品置于紫外灯光下连续照射0、0.5、1、2、3、6 h 后取出,分别取适量溶液用乙醇稀释20 倍测量紫草素吸光度,通过计算紫草素的保留率,考察加热不同时间以及紫外光照射不同时间对紫草素脂质纳米粒子的影响。计算紫草素保留率公式如下:
式中:CH(L)表示样品加热(或紫外光照射)后紫草素吸光度对应的浓度,mg/mL;C0表示初始样品紫草素吸光度对应的浓度,mg/mL。
1.2.3.2 pH 和离子稳定性 制备一系列不同pH 的磷酸缓冲溶液,将样品添加至不同pH 缓冲溶液中,使最终溶液pH 分别为2、3、4、5、6、7,在室温下平衡2 h,通过测量样品粒径大小,考察pH 对紫草素脂质纳米粒子的影响。制备一系列不同离子浓度的氯化钠溶液,将样品添加至不同离子浓度氯化钠溶液中,使最终离子浓度分别为0、100、200、500、800、1000 mmol/L,在室温下平衡2 h,通过测量样品粒径大小,考察离子浓度对紫草素脂质纳米粒子的影响。
1.2.3.3 冻融稳定性 将样品置于-20 ℃冰箱中冷冻4 h 取出常温下溶解后继续放入冰箱冷冻,连续重复三次,冷冻总时长为12 h。通过测量解冻后样品粒径大小和包封率来考察反复冻融对紫草素脂质纳米粒子的影响。取解冻后样品于8000 r/min 离心10 min 测量紫草素吸光度。计算紫草素包封率公式如下:
式中:CF表示冻融样品离心后紫草素吸光度对应的浓度,mg/mL;C0表示初始样品紫草素吸光度对应的浓度,mg/mL。
1.2.3.4 贮藏稳定性 将样品置于室温中贮藏4 周,通过测量样品粒径大小和保留率来考察贮藏时间对紫草素脂质纳米粒子的影响。每周定时取适量溶液测量样品粒径大小以及8000 r/min 离心10 min 测量紫草素吸光度。计算紫草素包封率公式如下:
式中:CS表示贮藏样品离心后紫草素吸光度对应的浓度,mg/mL;C0表示初始样品紫草素吸光度对应的浓度,mg/mL。
1.2.4 体外消化 静态模拟胃肠道消化实验可以考察紫草素经过消化被人体吸收利用情况。具体过程参考了之前对INFOGEST 方法进行的改进[27,33]。模拟口腔消化:将含有3 mg/mL 黏膜蛋白的口腔模拟液预热至37 ℃,然后与紫草素脂质纳米粒子按1:1体积比混合(7.5 mL+7.5 mL)。然后将混合物调至pH6.8,并在37 ℃下以100 r/min 的转速在振荡培养箱中放置10 min。模拟胃消化:将含有3.2 mg/mL胃蛋白酶的模拟胃液提前45 min 放入37 ℃水浴锅中活化。与通过口腔阶段的样品按1:1 体积比混合(15 mL+15 mL),将混合物调至pH 2.5,并在37 ℃下以100 r/min 的转速在振荡培养箱中放置2 h。模拟小肠消化:将模拟肠液与脂肪酶(24 mg/mL)、胰酶(24 mg/mL)、胆盐(54 mg/mL)溶于提前配好的5 mmol/L PBS(pH7.0)中,提前1 h 放入37 ℃水浴锅中活化。将通过胃阶段的样品调pH 至7.0 后加入1.5 mL 肠液,3.5 mL 胆盐,2.5 mL 胰酶和2.5 mL脂肪酶。将混合物pH 再一次调至7.0,在37 ℃下以100 r/min 的转速在振荡培养箱中放置2 h,期间要将pH 保持在7.0。体外消化实验模拟了样品依次经过口腔、胃、肠三个消化阶段后变成食糜,在此过程中经过酸、碱和酶的作用会损失一部分紫草素,食糜在4 ℃下经过15000 r/min 离心30 min 后得到的上清液被认为是包含了人体可吸收的紫草素的混合胶束部分。分别测量初始样品、食糜中和胶束中紫草素吸光度。计算紫草素保留率和生物可接受率公式如下:
式中:CC表示食糜中紫草素吸光度对应的浓度,mg/mL;C0表示初始样品紫草素吸光度对应的浓度,mg/mL;CM表示胶束中紫草素吸光度对应的浓度,mg/mL。
1.3 数据处理
所有实验至少重复3 次,最终结果以平均值±标准差的形式呈现,采用统计学分析软件SPSS Statistics 20 对数据进行分析,通过单因素方差和t检验分析数据间是否存在显著性差异,不同字母表示存在显著性差异(P<0.05)。
2 结果与分析
2.1 紫草素脂质纳米粒子的构建
如图1 所示,当不添加天然表面活性剂时,紫草素浓度从0.1 mg/mL 升至0.6 mg/mL 时,SLNP 的包封率由98.86%突降至47.20%。据报道[25],表面活性剂具有稳定纳米粒子的作用,推测在磷脂中添加表面活性剂能提高紫草素脂质纳米粒子的包封率和负载量,因此选取了两种天然表面活性剂茶皂素和槐糖脂。当紫草素浓度升至0.8 mg/mL 时,Sap-SLNP和Sop-SLNP 的包封率仍保持在95%左右。三种紫草素脂质纳米粒子都在0.4 mg/mL 处具有一个拐点,室温放置3 d 后包封率仍在0.4 mg/mL 处明显下降,此时纳米粒子中紫草素浓度为稳定状态的最高值。因此确定紫草素浓度为0.4 mg/mL。
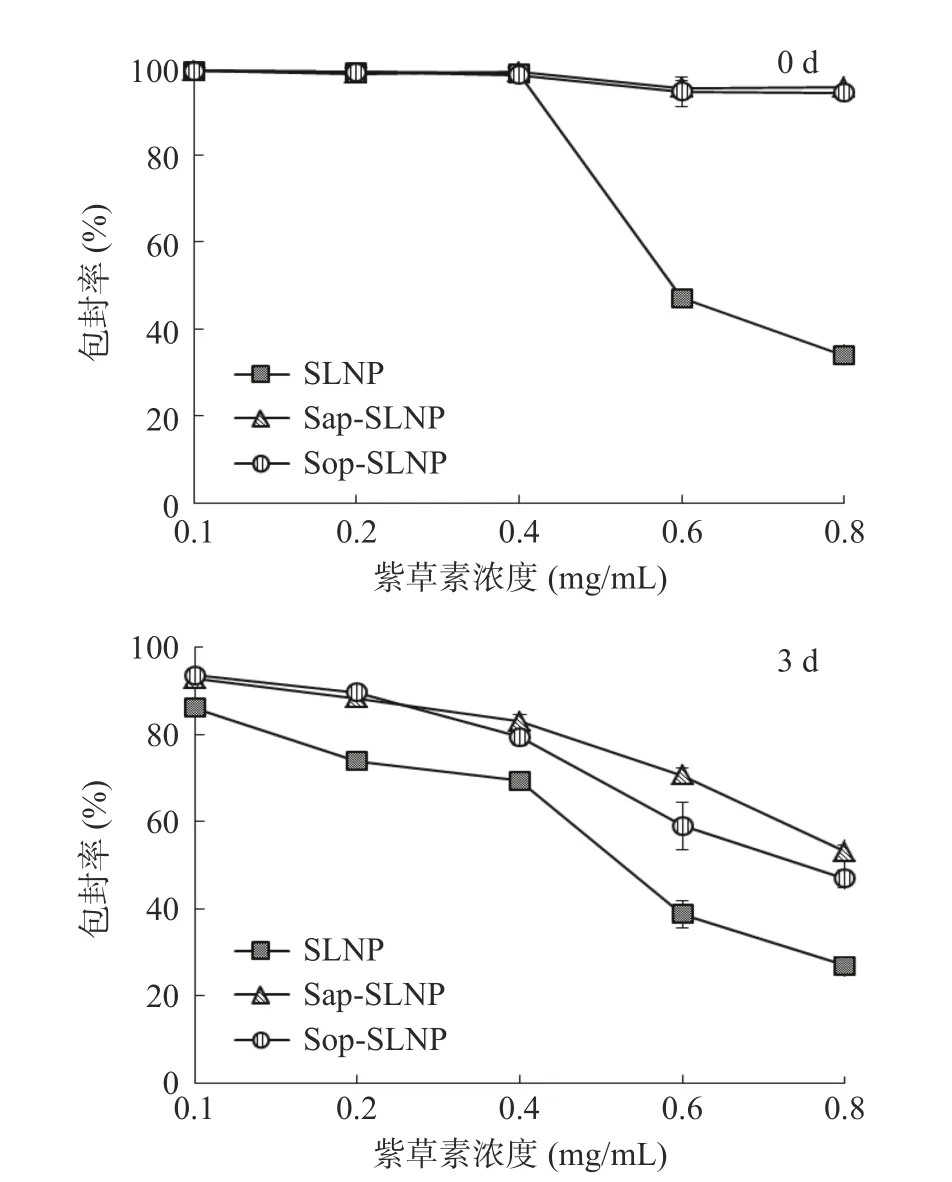
图1 紫草素浓度对紫草素脂质纳米粒子包封率的影响Fig.1 Effect of concentration of shikonin on encapsulation rate of shikonin lipid nanoparticles
如图2 所示,天然表面活性剂质量分数达到0.5%及以上时,脂质纳米粒子包封率由47%可提高到98%以上,并且室温放置3 d 后依然具有较高水平;在负载率方面,天然表面活性剂添加量为0.5%时具有最高值,由2.8%提高到5%以上。随着天然表面活性剂浓度增加至1%,负载率反而下降,说明此时几乎全部紫草素都已被包埋进脂质纳米粒子中,增加天然表面活性剂含量降低了紫草素在纳米粒子中的比例,使负载率减小。因此选择添加0.5%天然表面活性剂作为构建紫草素脂质纳米粒子的原料。
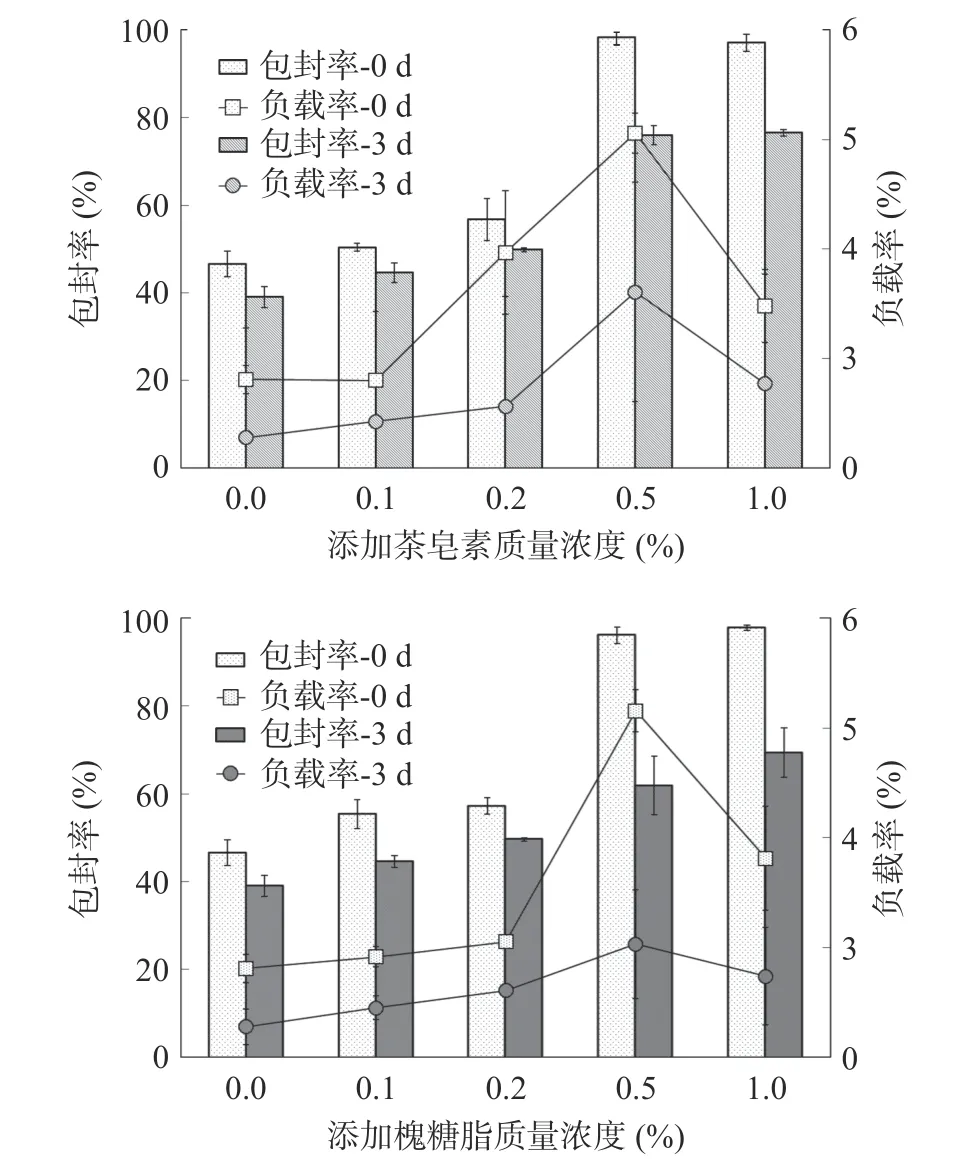
图2 添加表面活性剂对紫草素脂质纳米粒子包封率和负载率的影响Fig.2 Effects of surfactant addition on the encapsulation rate and loading rate of shikonin lipid nanoparticles
综合可得,紫草素脂质纳米粒子最终配方为卵磷脂(1%,w/v)和天然表面活性剂(茶皂素或槐糖脂)(0.5%,w/v),紫草素(0.4 mg/mL),此时Sap-SLNP最大包封率为99.01%±0.75%,最高负载率为5.06%±0.18%,Sop-SLNP 最大包封率为98.36%±0.30%,最高负载率为5.16%±0.19%。茶皂素(或槐糖脂)的加入大大提高了紫草素脂质纳米粒子的包封和负载能力,一方面是茶皂素(或槐糖脂)具有双亲性,其本身具有较好的吸附疏水性物质的能力[26-27],另一方面可能是茶皂素(或槐糖脂)与卵磷脂结合参与脂质体膜的形成,使磷脂分子排列更紧密,能容纳下更多紫草素而不会泄漏出去。有研究表明,皂苷有类似胆固醇的性质,替代脂质体中的胆固醇后,仍保持脂质体的包封率和稳定性[34]。Singh 等[35]的实验也表明槐糖脂能与脂质体膜结合。
2.2 紫草素脂质纳米粒子的结构表征
2.2.1 粒径与电位 通过动态光散射电泳仪测定了新制备的紫草素脂质纳米粒子的平均粒径、粒径分布和Zeta 电位,具体结果见表1。三种纳米粒子平均粒径为65 nm 左右,粒径分布集中,因此溶液呈现出澄清状态有较高的透光率。添加天然表面活性剂的粒子粒径没有明显增大,可能是因为包埋材料均通过了微射流处理达到了相同的纳米级别。电位为负值表示紫草素脂质纳米粒子带有负电荷,一般情况下,电位绝对值越大,带有相同电荷粒子之间的静电斥力越大,纳米粒子越不容易发生聚集沉淀,有利于维持体系的稳定[21]。Sap-SLNP 和Sop-SLNP 的电位绝对值均高于SLNP,这是因为添加天然表面活性剂增加了粒子所带负电荷,也侧面反映出Sap-SLNP和Sop-SLNP 具有更好的稳定性。
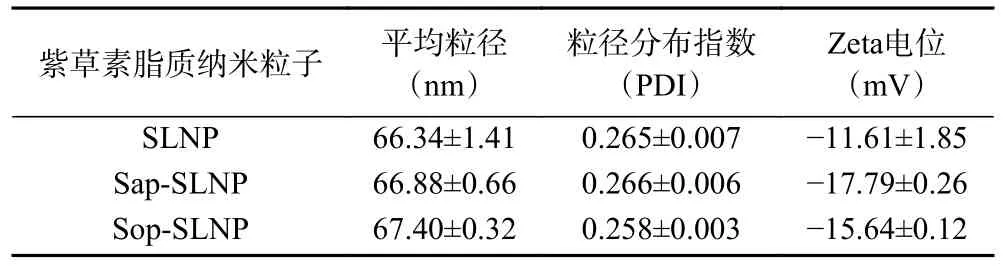
表1 紫草素脂质纳米粒子平均粒径、粒径分布指数和Zeta 电位Table 1 Mean particle size,particle size distribution and Zeta potential of shikonin lipid nanoparticles
2.2.2 原子力电子显微镜观察形态 通过原子力电子显微镜(AFM)观测了紫草素脂质纳米粒子的微观形貌。如图3 所示,三种紫草素脂质纳米粒子尺寸大小约为50~100 nm,与动态光散射仪测量结果一致,均呈现出表面光滑,分布均匀的球状结构,与脂质体的形态相似。其中,SLNP 呈现出更为圆滑的形状,而Sap-SLNP 和Sop-SLNP 更为尖细,这可能是茶皂素(或槐糖脂)与卵磷脂结合产生的相互作用力使磷脂圆球状结构发生了变化,纳米粒子整体变得更加紧密[36]。
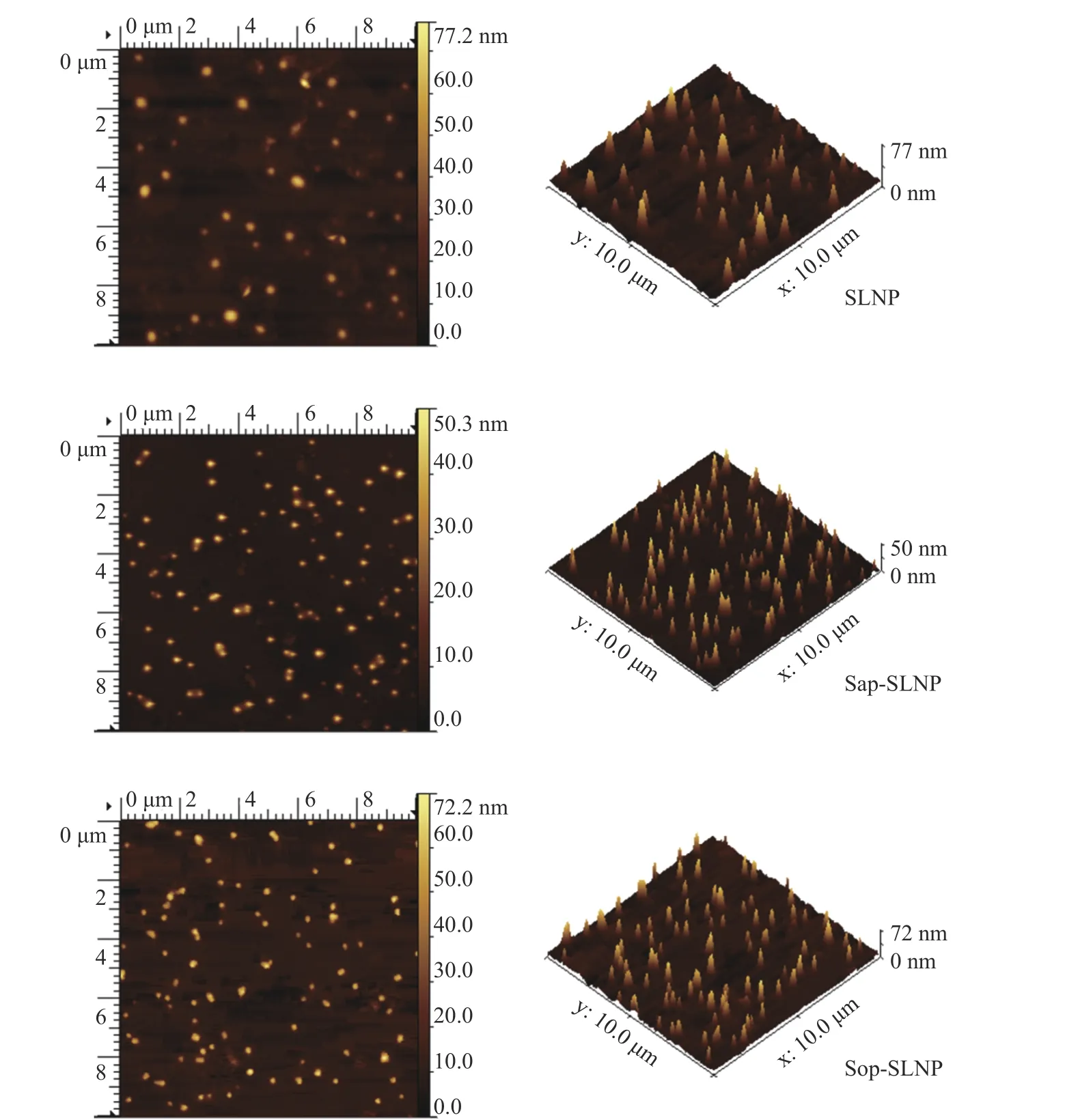
图3 紫草素脂质纳米粒子的AFM 图像Fig.3 AFM images of shikonin lipid nanoparticles
2.2.3 X 射线衍射(XRD)通过X 射线衍射法获得了紫草素脂质纳米粒子在2θ值(5°~90°)下的晶体衍射图。如图4 所示,紫草素在2θ衍射角5°~30°范围内结晶度高,衍射峰尖锐,而单独的卵磷脂以及卵磷脂与茶皂素,卵磷脂与槐糖脂在20°的衍射角上只有一个平坦的峰,说明这些原料主要以无定形状态存在。此外,单独的卵磷脂在2θ衍射角5°~10°范围内有尖锐的衍射峰,说明卵磷脂中有晶粒存在,可能是在冻干样品过程中,由于温度低于卵磷脂相变温度导致卵磷脂向结晶状态转变,而卵磷脂与茶皂素,卵磷脂与槐糖脂,在此范围的衍射峰相对强度降低,说明添加茶皂素(或槐糖脂)能抑制卵磷脂晶粒的形成。将紫草素包埋到纳米粒子中后,紫草素的特征结晶峰消失了,说明将紫草素包埋在纳米粒子疏水区抑制了紫草素的结晶,使紫草素以非晶态形式存在。有研究报道非晶态口服的生物利用度高于晶态口服生物利用度[37],为开发功能性食品提供了有利条件。此外,在衍射角30°~85°范围内有规则的结晶峰,这些峰是在制备样品过程中调pH 时由NaOH 和HCl 中和产生的NaCl 晶体的特征峰。
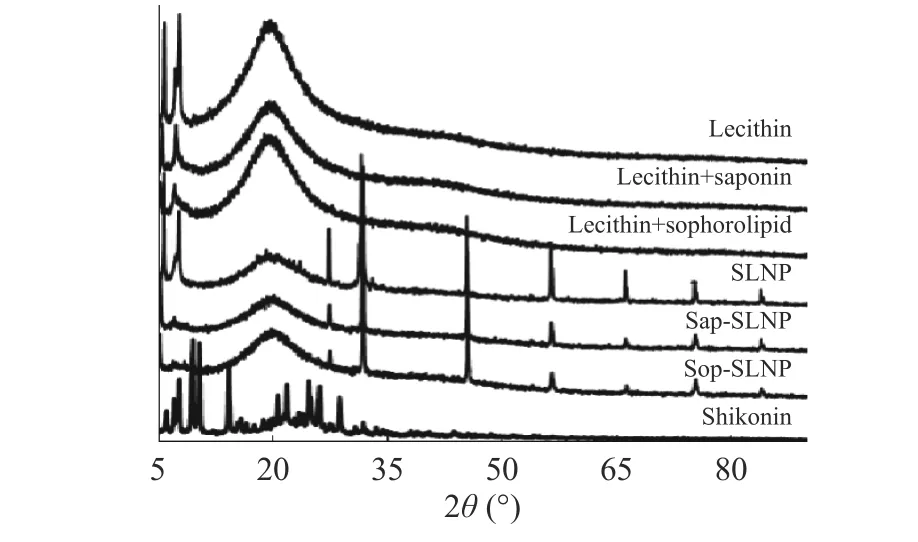
图4 紫草素、卵磷脂和天然表面活性剂以及紫草素脂质纳米粒子的XRD 图Fig.4 XRD patterns of shikonin,lecithin and natural surfactant and shikonin lipid nanoparticles
2.2.4 傅立叶变换红外光谱(FTIR)通过红外光谱分析紫草素与卵磷脂和天然表面活性剂之间的分子相互作用。如图5 所示,紫草素的特征峰出现在3252、2920、1610、1447、1342、1204、1066、779 cm-1,分别为O-H 氢键、烷基C-H、C=O、芳环和C-O 的拉伸振动[8]。在卵磷脂、卵磷脂与茶皂素(或槐糖脂)的FTIR 光谱中,在3400~3200 cm-1,均有一个宽峰,是O-H 氢键拉伸振动的表现;2926 和2857 cm-1两处峰是烷基CH2对称与不对称拉伸,1737 cm-1处的尖峰是卵磷脂-C=O 拉伸振动产生的;1247 cm-1附近的峰值对应了磷脂中PO4磷酸基团。与单独卵磷脂相比,添加了茶皂素(或槐糖脂)的FTIR 光谱中,在1608 和1064 cm-1附近出现了两个糖类的特征峰,这与茶皂素(或槐糖脂)结构中带有糖基有关;同时,1247 cm-1处的峰发生红移,OH 基团峰的强度和范围也发生了改变,说明茶皂素(或槐糖脂)与卵磷脂中的磷酸基团发生了相互作用[24],可能是糖羟基与磷酸基团产生了分子间氢键[23]。加入紫草素形成纳米粒子后紫草素脂质纳米粒子光谱变化不大,说明紫草素与卵磷脂和天然表面活性剂之间没有形成共价键,只有O-H 氢键波段部分有移动,说明紫草素与卵磷脂和天然表面活性剂之间形成了氢键[9]。而紫草素在1610~779 cm-1处特征峰消失了说明包埋进纳米粒子后紫草素的振动拉伸和弯曲受到了限制[22]。
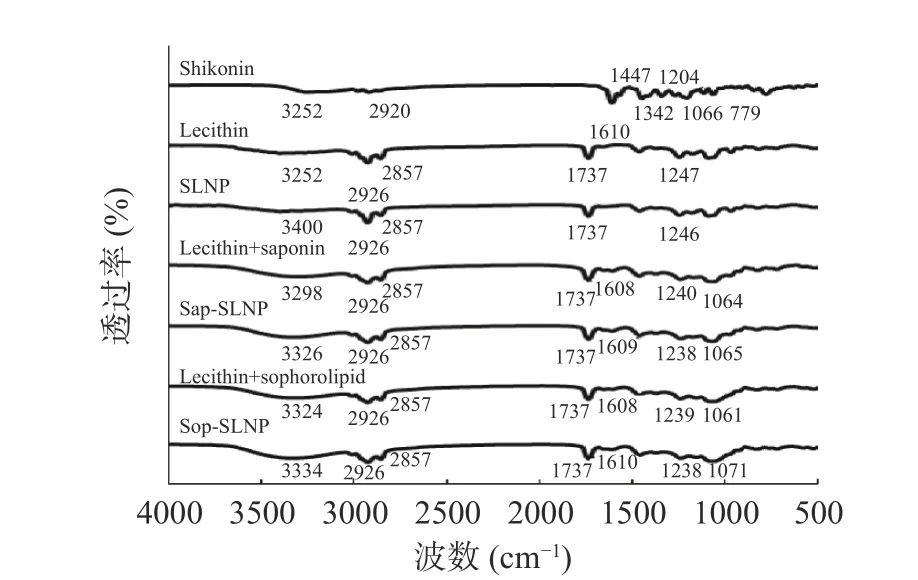
图5 紫草素、卵磷脂和天然表面活性剂以及紫草素脂质纳米粒子的FTIR 光谱图Fig.5 FTIR spectras of shikonin,lecithin and natural surfactant and shikonin lipid nanoparticles
2.3 紫草素脂质纳米粒子的稳定性
2.3.1 热和光稳定性 紫草素易受热和光分解,如图6(A、B)所示,分别经过加热和光照处理,游离紫草素保留率仅40.92%±6.53% 和53.44%±7.95%,SLNP 的保留率为69.33%±3.04%和77.53%±4.24%,说明脂质纳米粒子包埋紫草素起到了一定的保护作用,而Sap-SLNP 和Sop-SLNP 的保留率均在此基础上进一步提高,热保留率分别为77.25%±2.29%、80.08%±2.60%,约是游离紫草素的2 倍,光保留率为83.39%±3.75%、82.31%±4.44%,约是游离紫草素的1.6 倍。这说明,添加茶皂素(或槐糖脂)能进一步提高紫草素脂质纳米粒子的热和光稳定性,根据AFM 图像可能是因为茶皂素(或槐糖脂)的加入使磷脂分子排列更加紧密,粒子整体结构更加紧密[36],减少了磷脂分子因受热和光氧化而发生断裂。
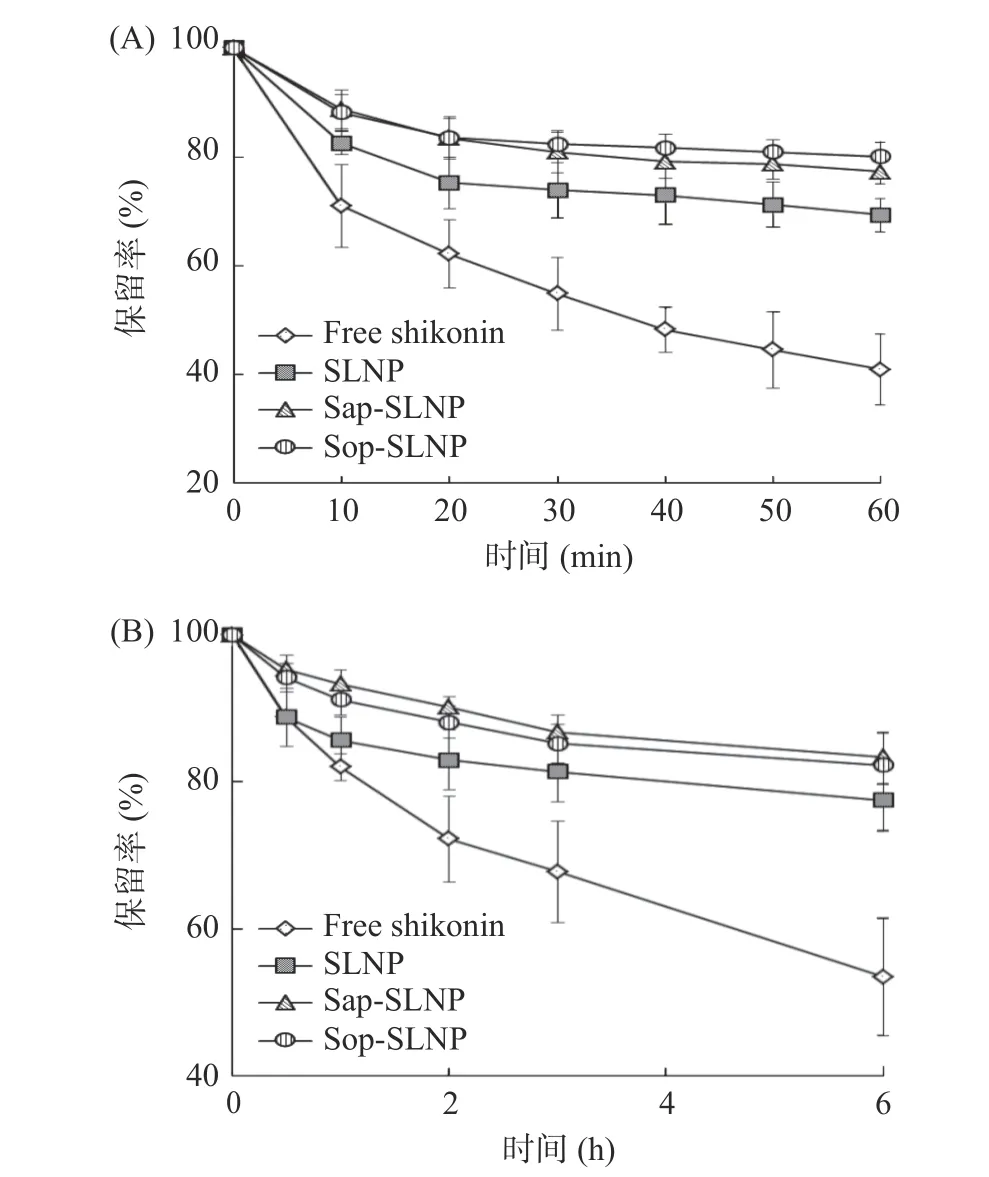
图6 加热(A)和紫外光照(B)时间对紫草素脂质纳米粒子保留率的影响Fig.6 Effects of heating (A) and ultraviolet illumination (B)on the retention rate of shikonin lipid nanoparticles
2.3.2 pH 和离子稳定性 pH 和离子稳定性考察脂质纳米粒子在不同环境及加工条件下的稳定性。如图7(A)所示,在溶液pH2~7 环境下,SLNP 粒径在pH 5 及以下逐渐变大,这可能是由于溶液中H+增加使粒子间静电力减小,从而使纳米粒子更易聚集,而Sap-SLNP 和Sop-SLNP 粒径大小并未发生明显变化,表明Sap-SLNP 和Sop-SLNP 具有良好的pH 稳定性。如图7(B)所示,在氯化钠离子浓度0~1000 mmol/L溶液中,SLNP、Sap-SLNP 和Sop-SLNP 粒径大小均并未发生明显变化。结果表明紫草素脂质纳米粒子具有良好的pH 和离子稳定性,有利于在不同加工条件下生产。
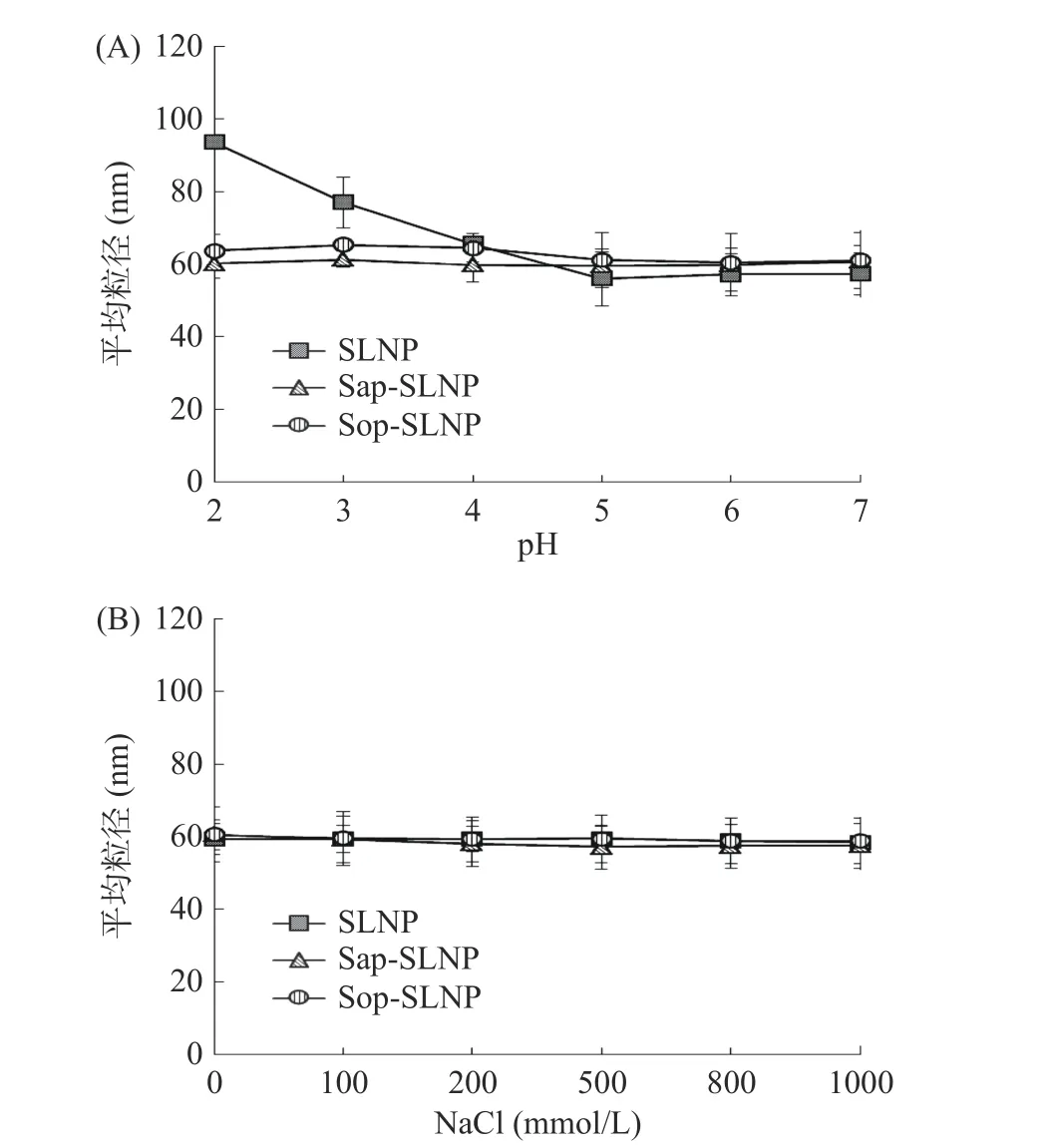
图7 pH(A)和离子浓度(B)对紫草素脂质纳米粒子粒径大小的影响Fig.7 Effects of pH (A) and ion concentration (B) on the particle size of shikonin lipid nanoparticles
2.3.3 冻融稳定性 在反复冻融过程中,脂质体中的水分子会结成冰晶,对脂质体膜造成损伤,破坏脂质纳米粒子的结构导致内容物泄漏[38],因此冻融稳定性从包封率与粒径大小变化两个方面表征。经过12 h连续三次反复冻融后,SLNP 包封率仅为46.23%±1.42%,这是由于水分子结晶导致磷脂膜破损,紫草素泄漏出来从而降低了包封率,而Sap-SLNP 和Sop-SLNP 包封率分别为87.28%±4.13%、80.95%±7.55%,将近SLNP 的2 倍,说明反复冻融对Sap-SLNP 和Sop-SLNP 结构的破坏损伤小,可能是因为茶皂素(或槐糖脂)含有糖残基,抑制了水分子的结晶[39]。SLNP 粒径由(59.35±4.41)nm 增大至(543.38±90.21)nm,而Sap-SLNP 和Sop-SLNP 粒径分别增大至(101.60±12.60)nm、(148.06±37.07)nm,由图8可见,经过冻融后,SLNP 明显变浑浊,而Sap-SLNP和Sop-SLNP 较为清澈,Sap-SLNP 效果更好,与粒径大小结果一致。这可能是因为茶皂素(或槐糖脂)在冻融过程中起到了保护脂质纳米粒子结构的作用[40],使纳米粒子在这个过程中不易被破坏和聚集。
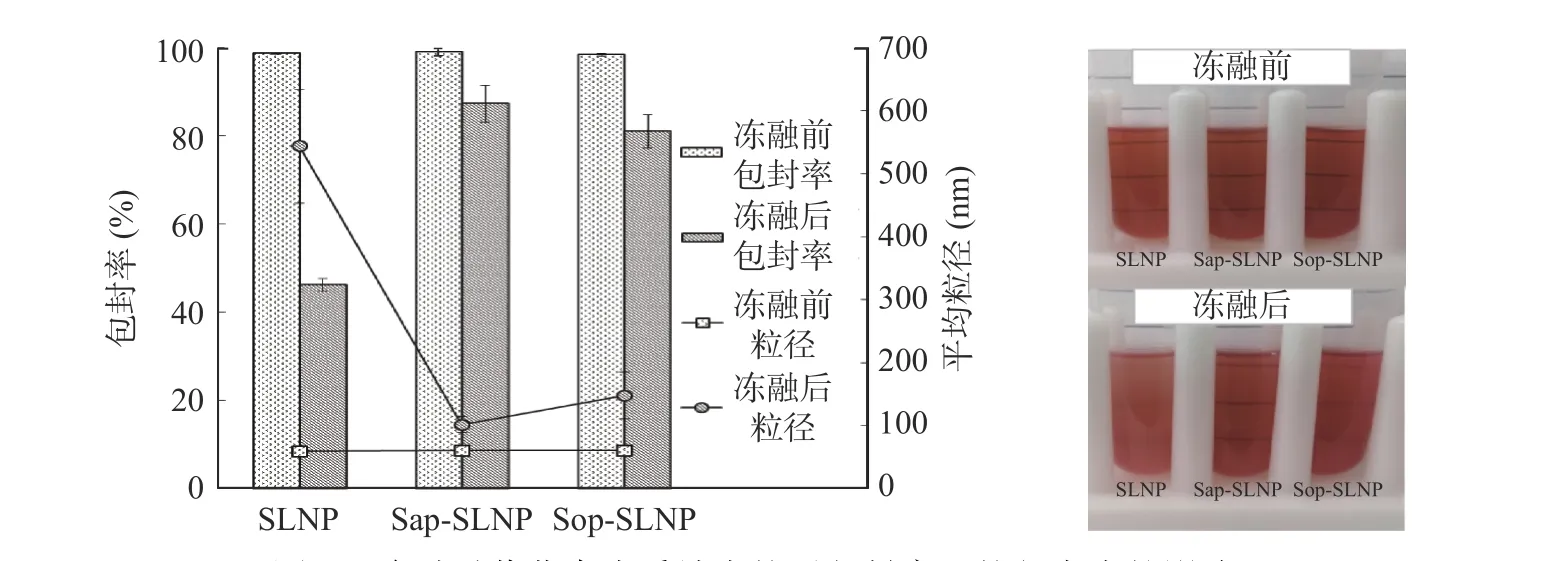
图8 冻融对紫草素脂质纳米粒子包封率和粒径大小的影响Fig.8 Effect of freezing-thawing on the encapsulation efficiency and particle size of shikonin lipid nanoparticles
2.3.4 贮藏稳定性 脂质体在贮藏过程中易降解、聚集和融合导致包埋物质的泄漏[24]。因此通过检测紫草素脂质纳米粒子在贮藏过程中包封率和粒径大小来评估其贮藏稳定性。如图9(A),贮藏4 周后,SLNP 包封率仅为48.38%±1.76%,Sap-SLNP 和Sop-SLNP 的包封率分别降至80.25%±0.78%、74.03%±3.55%,约是SLNP 的1.7 倍,第3 周至第4 周,包封率趋于平稳。由图9(B)可知,SLNP 粒径随着时间增长而变大,这是因为在贮藏过程中磷脂发生聚集导致粒径变大;而Sap-SLNP 和Sop-SLNP 在贮藏过程中一直保持稳定的粒径大小,这可能与Sap-SLNP和Sop-SLNP 粒子本身之间产生的较大的静电斥力有关[21]。结果表明,Sap-SLNP 和Sop-SLNP 表现出良好的贮藏稳定性,茶皂素(或槐糖脂)的加入能使紫草素脂质纳米粒子状态更加稳定,延长贮藏时间。
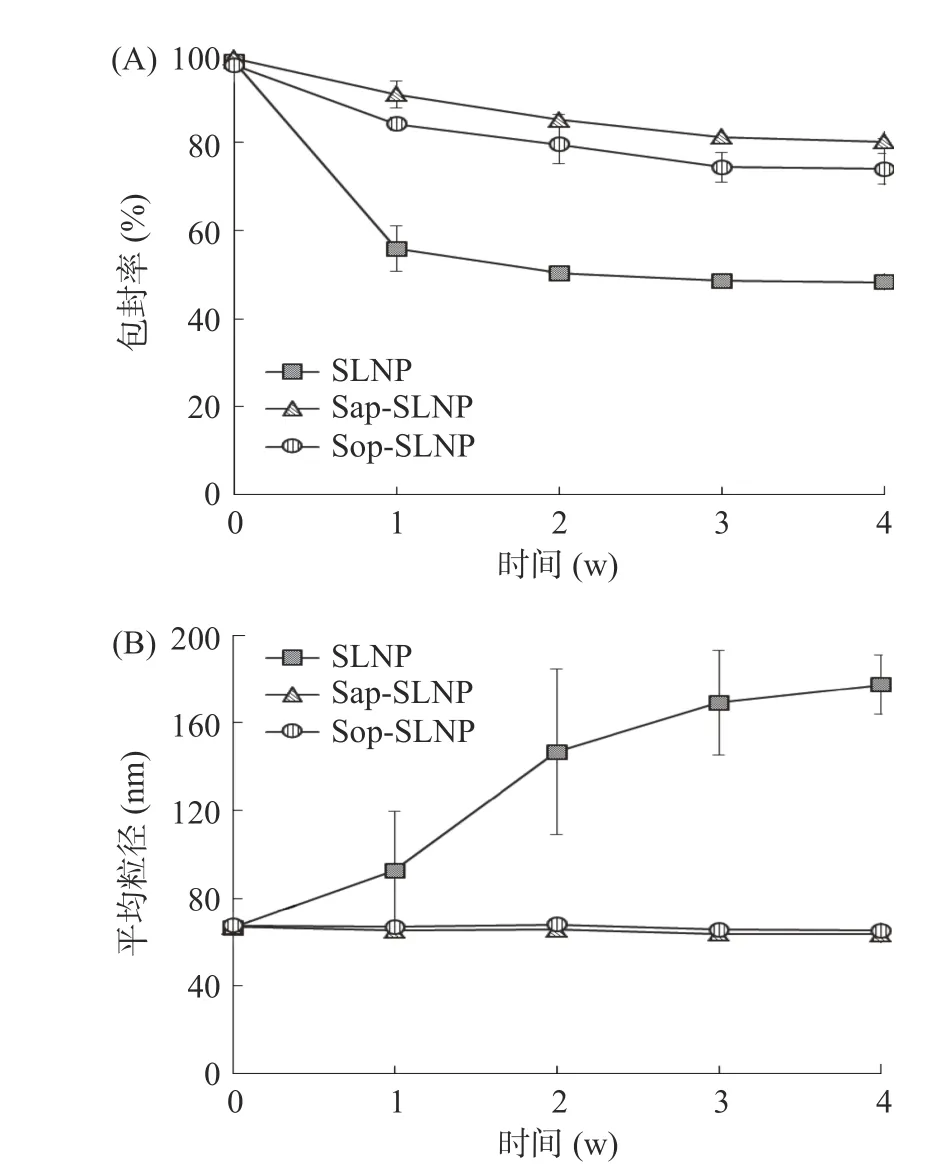
图9 贮藏时间对紫草素脂质纳米粒子包封率和粒径大小的影响Fig.9 Effect of storage time on encapsulation efficiency and particle size of shikonin lipid nanoparticles
2.4 紫草素脂质纳米粒子的体外消化
紫草素是一种疏水性多酚,因此具有很低的水溶解性,从而导致其生物利用率低。通过保留率和生物可接受率来评估活性物质的体外消化结果。保留率指生物活性物质经过模拟消化后体系中剩余的量[27,41]。紫草素的降解主要是由暴露在中性或碱性水环境引起的,因此通过防止紫草素与周围的水相接触来减少降解[28]。由于游离紫草素以晶体形式存在,而纳米粒子的尺寸小得多,暴露在水相中的紫草素的表面积要比自由晶体大得多,因此纳米粒子中的紫草素可能比较大,晶体中的紫草素更容易降解。如图10所示,游离紫草素的保留率为86.89%±2.03%,高于紫草素脂质纳米粒子,紫草素脂质纳米粒子之间保留率相差不大,分别为58.96%±1.72%,55.15%±1.27%和55.67%±1.51%,这可能与纳米粒子粒径尺寸相同有关。生物可接受率指生物活性物质经过模拟消化后溶解在胶束中可用于吸收的比例[27,42]。游离紫草素的生物可接受率仅15.94%±0.85%,通过包埋技术,SLNP 的生物可接受率大幅度提高至42.56%±0.92 %,Sap-SLNP 和Sop-SLNP 的生物可接受率在此基础上进一步提高至49.15%±1.41%和48.29%±0.66%,将紫草素生物可接受率分别提高到3.08 倍和3.03 倍。一方面是由于经过纳米粒子包埋,表面积增加导致紫草素比较大的晶体更易溶解;另一方面,纳米粒子中的紫草素是无定形状态的,比结晶形式更容易溶解。因此利用包埋技术能有效提高紫草素在消化过程中的溶解度,从而达到更易吸收的效果。添加茶皂素(或槐糖脂)后,能进一步提高紫草素的生物可接受率,可能是因为紫草素在单纯磷脂中是比较集中存在于磷脂双分子层的疏水区,而加入的茶皂素(或槐糖脂)可以通过与磷脂结合或者吸附于纳米粒子表面增大了紫草素分布范围,使紫草素无定形形态更加松散,从而更易溶解于水中[27,41]。
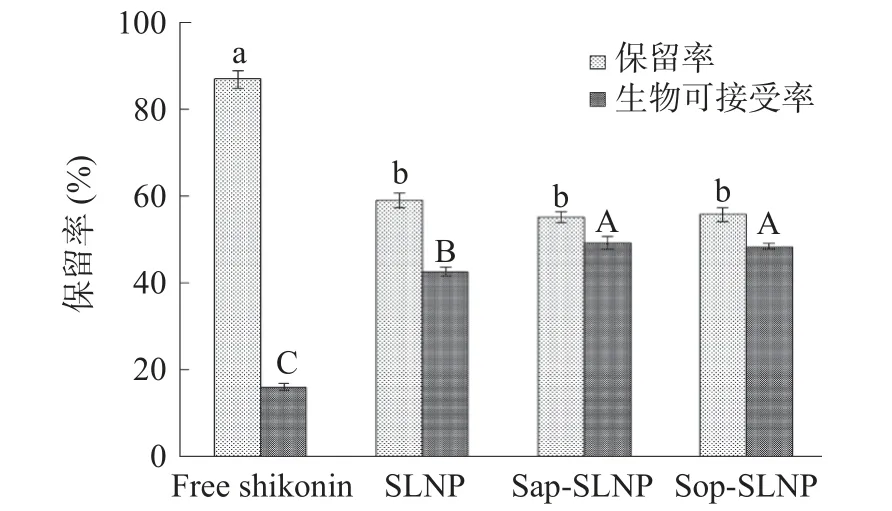
图10 紫草素脂质纳米粒子经过体外消化后的保留率与生物可利用率Fig.10 Retention rate and bioaccessibility of shikonin lipid nanoparticles after in vitro digestion
3 结论
本课题采用卵磷脂添加茶皂素或槐糖脂构建紫草素脂质纳米粒子,成功制备出了两种天然表面活性剂修饰的紫草素脂质纳米粒子。结果表明,与单纯卵磷脂包埋的紫草素脂质纳米粒子相比,将卵磷脂和天然表面活性剂(茶皂素和槐糖脂)质量比为2:1 混合制备的紫草素脂质纳米粒子能显著提高脂质纳米粒子的包封率(98%以上)和负载率(5%以上),且进一步提高了紫草素脂质纳米粒子的环境稳定性。两种天然表面活性剂修饰的紫草素脂质纳米粒子环境稳定性相近,热稳定性和光稳定性分别约是游离紫草素的2 倍和1.6 倍,在pH2~7 以及离子浓度1000 mmol/L内依然保持较好的稳定性,经过三次反复冻融后粒径变化不大,包封率仍有85%左右,将近是单纯脂质纳米粒子的2 倍,并且经过4 周储藏后粒径基本不变,包封率仍有80%左右,约为单纯脂质纳米粒子的1.7 倍,并且显著提高了紫草素的生物利用率,约是游离紫草素的3 倍。这些研究结果为紫草素的进一步开发利用提供了良好的思路和理论指导,对提高紫草素在功能性食品、膳食补充剂或药物制剂中的稳定性和生物利用度具有重要意义,并可能促进天然胶体递送系统的发展。
猜你喜欢
农药科学与管理(2019年10期)2019-04-20
特别健康(2018年9期)2018-07-17
中成药(2018年6期)2018-07-11
天然产物研究与开发(2018年1期)2018-02-02
广东饲料(2016年7期)2016-12-01
中国粮油学报(2016年1期)2016-02-06
家教世界·创新阅读(2015年12期)2015-12-10
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年10期)2015-03-01
云南中医学院学报(2014年6期)2014-07-31
