
广西凤山中籽茶群体种质SNP标记开发及遗传进化分析
2024-01-30 17:32付超刘凯张幸梁国校江泽鹏王凌晖
经济林研究 2023年3期
关键词:油茶
付超 刘凯 张幸 梁国校 江泽鹏 王凌晖
摘 要:【目的】開发大量SNP 标记用于油茶种质资源鉴定及其遗传差异化分析,为广西优良地方油茶种质资源的开发和利用提供依据。【方法】以广西凤山中籽茶8 个居群64 份种质资源的叶片为供试材料,参考已公布的茶树Camellia sinensis var.sinensis 基因组进行酶切预测,选择最适酶切方案,对油茶基因组DNA 酶切构建SLAF-seq 文库,利用SLAF-seq 测序技术对其进行高通量测序、SNP 标记开发及其进化关系分析。【结果】通过对照组水稻(Oryza sativa)测序数据的评估检测,显示本次试验双端比对效率为95.53%,酶切效率为95.06%,表明SLAF 建库正常;构建SLAF-seq 文库并进行双端测序,平均测序深度为13.43 x,共获得207.34 M 的读长数据,测序平均Q30 为88.94%,平均GC 含量为44.85%,共开发879 284 个SLAF 标签,其中多态性SLAF 标签242 892 个,共得到4 565 217 个群体SNP,进一步地开发获得199 745 个高一致性的SNP 标记;基于高一致性SNP 标记的遗传分析结果表明:可将这广西凤山中籽茶8 个居群64 份种质划分为2 个大类群,4个亚类群,每个亚类群均由来源于1 ~ 3 个地方群体种质组成。【结论】本研究表明SLAF-seq 测序技术可有效用于油茶群体种质SNP 标记开发,其标记数量、质量及效率远远优于ISSR、SRAP、SSR 等第一、二代分子标记技术;聚类分析表明,遗传变异多来自于群体内,群体间各群各自聚类、地域性相对明显,其遗传分化程度与地理距离远近呈现显著的正相关性。从全基因组范围开发的高质量SNP 标记可进一步用于油茶种质资源鉴定、连锁图谱构建以及重要性状的关联分析等研究。
关键词:油茶;SNP;SLAF-seq;遗传分析
中图分类号:S794.4 文献标志码:A 文章编号:1003—8981(2023)03—0169—07
油茶Camellia oleifera 别名茶油树、茶子树,是我国南方特有的优质食用油料树种。油茶籽油中不饱和脂肪酸含量高达80% 以上,因其组分与橄榄油相似,故有“东方橄榄油”美誉[1]。同时,油茶籽油富含生育酚、角鲨烯等天然抗氧化剂成分,且能够降低人体血清中甘油三酯含量而提高有益胆固醇含量,与其他植物油相比,是一种理想的营养保健食用油[2]。我国油茶栽培历史悠久,主要栽培区为湖南、江西、广西、湖北等地。目前,通过国家审定的油茶良种有百余个,如湖南“湘林”系列、“三华”系列,江西“长林”系列、“赣无”系列、广西“岑软”系列等[3]。油茶是异花授粉植物,遗传背景比较复杂,个体间变异性大,加上普通油茶常为多倍体,这给油茶种质资源遗传多样性分析、评价及利用带来了很大困难[4-5]。
群体遗传学是植物遗传学研究的重要组成单位。随着分子标记技术的快速发展,其在群体遗传学研究中也得以广泛应用,其原理为利用应用数学和统计学方法,分析全基因组中大量高准确的核苷酸变异信息,讨论群体遗传结构、物种形成机制、基因交流情况和群体进化动态等研究问题,阐述植物进化机制规律为育种工作提供理论基础[6-7]。同时,通过全基因组高密度核苷酸的多态性位点,研究种群间及群体内个体的基因交流、突变及自然选择等遗传进化过程影响机制,也提供了一种在基因水平上研究群体遗传学的新方法。SLAF-seq(Specific-Locus Amplified FragmentSequencing)是以高通量测序为基础的简化基因组测序技术。通过生物信息学进行实验方案系统设计,用特定的限制性内切酶对基因组进行酶切,筛选特异长度的DNA 片段(即SLAF 标签),构建SLAF-seq 文库,测序获得海量序列数据,利用软件进行数据信息分析,进而开发特异性的分子标记(SNP 和InDel)[8]。SLAF-seq 可在短时间内以较低的成本实现全基因组范围的分子标记开发,是目前最热门的分子标记开发技术之一。该技术具有通量高、有效reads 长、准确度高、重复性好、成本低等特点[9]。
凤山中籽茶C. oleifera of Fengshan,也称凤山中果茶,泛指广西凤山县境分布的有一定栽培历史的一类普通油茶实生林。凤山中籽茶农家种质作为广西油茶种质资源的重要组成部分之一,因其对当地栽培环境具有较好的适应性和抗逆性,加之其具有果实皮薄、籽粒大等优良经济性状,且适宜当地陡坡落地捡籽作业习惯,深得当地种植户青睐。但这一区域的油茶特色地方优良种质资源长期处于自然放纵状态,至今尚未得到有效研发和利用,长期缺乏地方良种支撑,一定程度制约了当地油茶产业的发展。应立足当地油茶资源,开展区域性特色地方种质遗传多样性研究,是充分发挥我区现有油茶优良种质资源优势,促进油茶产业良种化健康发展的需要,也是一项迫在眉睫的良种改造工程。本研究以广西凤山中籽茶8 个居群64 份种质为供试材料,采用SLAF-seq技术,获取大量多态性SLAF 标签,进而开发出一批高一致性的SNP 位点,并对凤山中籽茶开展遗传多样性分析,探索其遗传进化历程、生存状况,为下一步凤山中籽茶核心种质保护及优异形状选育利用等研究奠定基础。
1 材料与方法
1.1 材 料
本研究选用广西凤山中籽茶8 个居群64 份种质,分别为广西凤山县乔音、砦牙、长洲、金牙、中亭、平乐、江州、袍里等8 个乡镇的居群;从每个居群中分别选择8 个单株作为样株,样株选择隔离采样,每隔500 m 选择一株,分别采取无病虫害的嫩叶片,-70 ℃保存备用。每个品系按照1 ~ 8 进行编号(基本信息见表1)。
1.2 方 法
1.2.1 基因组DNA 提取及质量检测
采用改良CTAB 法提取供试64 份油茶种质的叶片gDNA,以琼脂糖凝胶电泳和紫外分光光度计分别检测DNA 的完整性、纯度及浓度,油茶gDNA 检测合格后,可供后续试验。
1.2.2 酶切建库及高通量测序
目前,尚未见到普通油茶全基因组序列信息公布,无法直接通过其基因组信息选择最优的限制性内切酶进行酶切,故需一个已公布基因组信息的油茶近缘物种作为参考基因组,通过在线电子酶软件(SLAF-predict)预测筛选出合适的限制性内切酶及酶切方案。茶树C. sinensis var.sinensis 是山茶科山茶属植物,为油茶近缘种,且已公布全基因组序列信息(http://tpia.teaplant.org/index.html,基因组大小为3.1 Gb,GC 含量为42.3%),故可选择茶树基因组作为酶切参考,筛选最适限制性内切酶及酶切方案,并对油茶基因组DNA 进行酶切。将获得的所有酶切片段(SLAF标签)进行3 末端加A 尾巴处理、连接专用测序接头、PCR 扩增、纯化、混样、切胶、回收目的片段,质检合格后的SLAF 文库用于IlluminaHiSeqTM 高通量测序。
为评估供试油茶酶切实验的准确性,选取以水稻O. sativa 作为对照同时进行测序检测验证。利用Dual-index 软件识别高通量测序获得的所有原始数据,得到各个样品所有的读长(reads),并过滤测序所有reads 的接头、低质量阅读框和去污染处理,进行碱基含量分布值(GC 含量)和测序质量值(Q30)分析,以此保证所有供试样品的测序质量及数据量。最后利用SOAP 软件[10] 将对照(水稻)的测序reads 与已公布的水稻组(http://rapdb.dna.affrc.go.jp/)进行比对,进行酶切效率评估,得以判断整个实验酶切测序过程的准确性和有效性。
1.2.3 油茶特异性SNP 位点的开发及其遗传分析
根据过滤去除获得的所有原始测序reads 序列相似性,将各样品测序产生的reads 分别进行聚类,所有供试样品中聚类到一起的reads 均来自于同一个SLAF 片段(SLAF 标签);不同样品间的同一个SLAF 标签在序列有碱基差异、缺失(即有多态性),即定义为多态性SLAF 标签。开发SNP 标记是以深度最高的每个SLAF 标签序列类型作为参考序列,利用BWA[11] 软件技术将测序得到的reads 比对到参考序列上,再使用GATK[12] 和samtools[13] 两种软件分析方法分别开发SNP,最终以两种方法得到的交集SNP 标记作为可靠的SNP标记。通过MEGA 6.0 软件,采用neighbor-joining算法,对所有供试样株进行遗传进化分析,构建系统发育树。同时,利用cluster 软件,设置第一、第二、第三主成分,进行主成分分析(Principalcomponent analysis,PCA),得到所有供试样品的主成分聚类信息。
2 结果与分析
2.1 建库评估
酶切成功与否是直接影响后续测序及分析实验成功的关键指标之一。利用SLAF-predict 软件,通过对茶树参考基因组进行电子酶切来预测,最终确定选取Hae Ⅲ为限制性内切酶;对酶切片段的进一步分析表明,获得的茶树基因组酶切片段基本上均匀分布其基因组上,且在重复序列区中的标签占比较少,并把筛选出长度在414 ~ 464 bp的酶切片段序列界定为SLAF 标签,预测共获得130 019 个SLAF 标签。为进一步评价酶切方案的可靠性与酶切效率,以评估监控水稻(对照)测序数据的实验过程正常与否,确定酶切方案实施的可靠性与酶切效率。通过SOAP 软件将水稻的测序reads 与已公布的水稻组(http://rapdb.dna.affrc.go.jp/)进行比对,结果显示,水稻双端比对效率为95.53%,酶切效率为95.06%,表明SLAF建库正常。
2.2 測序数据统计与评估
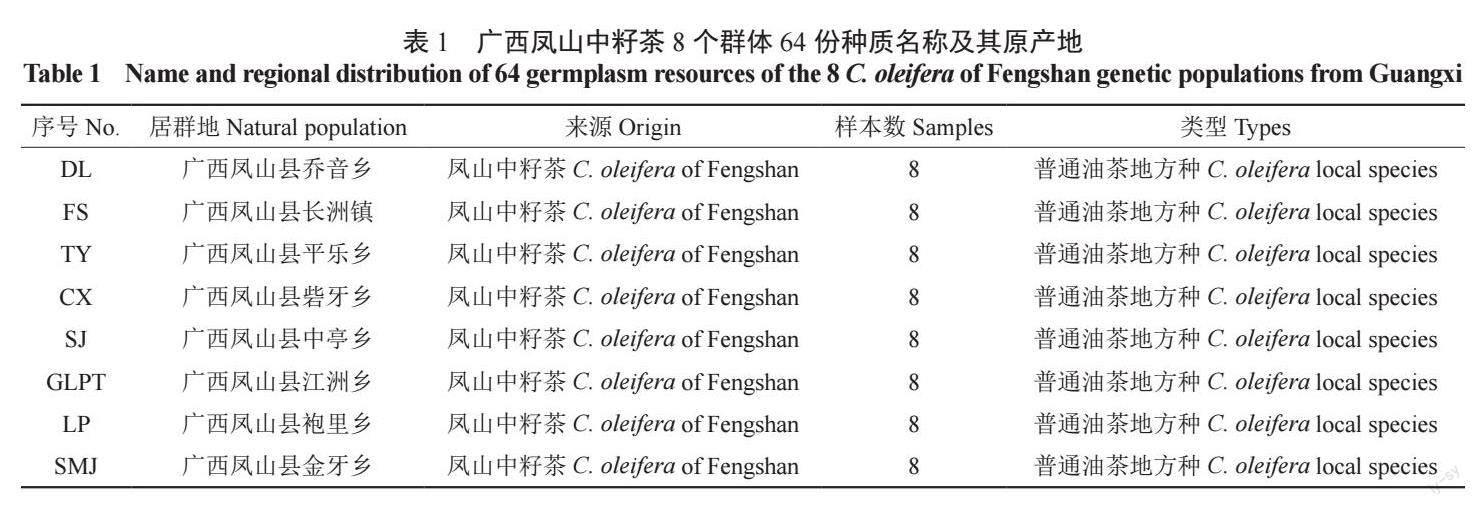
为保证数据分析质量,本研究采用读长(reads)125 bp×2 作为后续的数据评估和分析。经过Illumina HiSeqTM 高通量测序平台对所有供试样品64 份油茶及对照水稻进行测序,共获得207.34 Mreads 数据,对照水稻的数据量为0.15 M reads;64 份油茶样品测序获得的数据量在1.27 ~ 6.07 Mreads 之间,GLPT-3 获得的数据量最小为1.27M reads,SJ-7 获得的数据量最大为6.07 M reads(图1A)。GC 含量在43.24% ~ 46.43% 之间,所有供试样品测序的平均GC 含量为44.85%(图1B),说明达到了测序要求。
在测序识别过程中,每个碱基都会有1 个测序质量值(Q),其是衡量高通量测序中碱基识别是否正确的重要指标,以此便于衡量此碱基被识别的稳定性和准确性,该值越高表明碱基测序错误率越低;如果碱基的测序出错的概率为1‰,则其该碱基的质量值Q 为30,所以一般将测序质量值(Q)大于等于30(Q30)作为测序结果较为可靠的标准。本研究64 份油茶样本的Q30 在85.93% ~ 92.03% 之间,所有样品测序平均Q30达到88.94%(图1B),说明测序错误率较低,测序数据良好并可用于下一步分析。
2.3 SLAF 标签与特异性SNP 位点开发
所有供试样本共获得879 284 个SLAF 标签, 其中,242 892 个多态性SLAF 标签, 占开发SLAF 标签总数的27.6%,每个供试样品平均得到141 067 个SLAF 标签,样本平均测序深度为13.43 x,( 图1C—D)。采用GATK 和samtools两种软件分别开发SNP,以两种方法获得的交集SNP 标记作为最终可靠的SNP 标记,本研究共获得4 565 217 个群体SNP;SNP 的杂合率在10.81% ~ 33.51% 之间, 完整度在48.28% ~98.15% 之间;根据完整度> 0.8,最小等位基因频率(MAF)> 0.05 过滤,共得到199 745 个高一致性的广西凤山中籽茶群体SNP(图1E—F)。
2.4 遗传进化分析
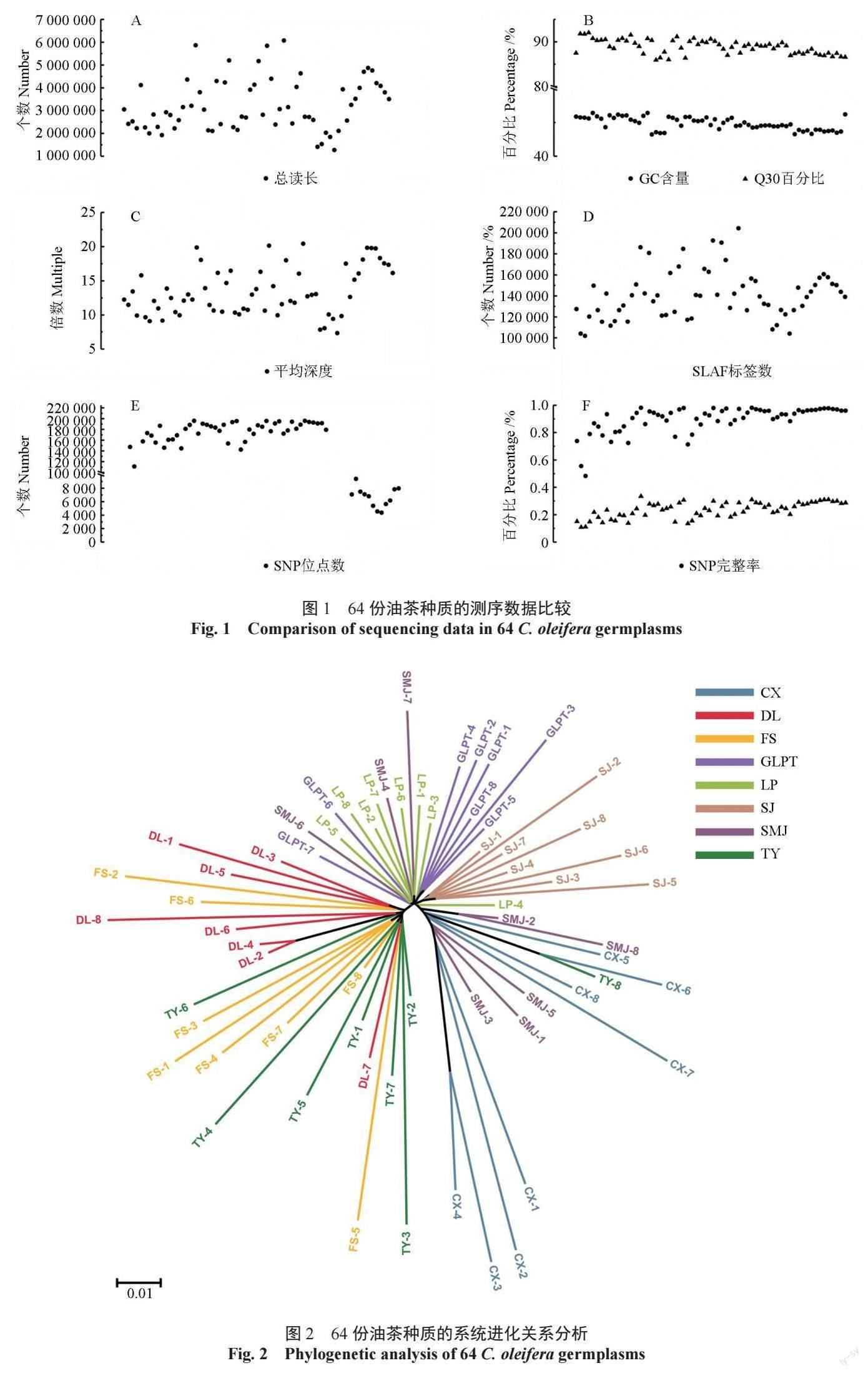
基于获得的199 745 个高一致性的广西凤山中籽茶群体SNP, 通过MEGA 6.0 软件, 采用neighbor-joining 算法,对广西凤山中籽茶8 个居群64 份种质进行系统进化关系分析,构建其进化树。聚类结果表明,可将64 份油茶种质划分为两个大类群,继续细分可分为A、B、C、D 这4 个亚类群(图2),第一大类群只包含A 亚类群,由广西凤山县乔音乡(DL)、长洲镇(FS)、砦牙乡(CX)3 个居群组成;第二大类群包含B、C、D 三个亚类群,由广西凤山县平乐乡(TY)、中亭乡(SJ)、江洲乡(GLPT)、袍里乡(LP)、金牙乡(SMI)5 个居群组成。亚群A、B 和D 中均在不同地理来源的种群,说明对应亚群内部的居群亲缘关系较近,但第一大类群(TY,DL,FS)中3 个居群和第二大类群5 个居群(CX,GLPT,LP,SJ,SMJ)的个体间无交叉聚类的现象,说明两个大类群的居群遗传关系较远。进一步结合居群地理信息发现,第一大类居群均来自广西凤山县西南部,第二大类居群均来自广西凤山县的东北部,由此可见,不同地理来源的居群与其遗传分化情况一致。
2.5 PCA 分析
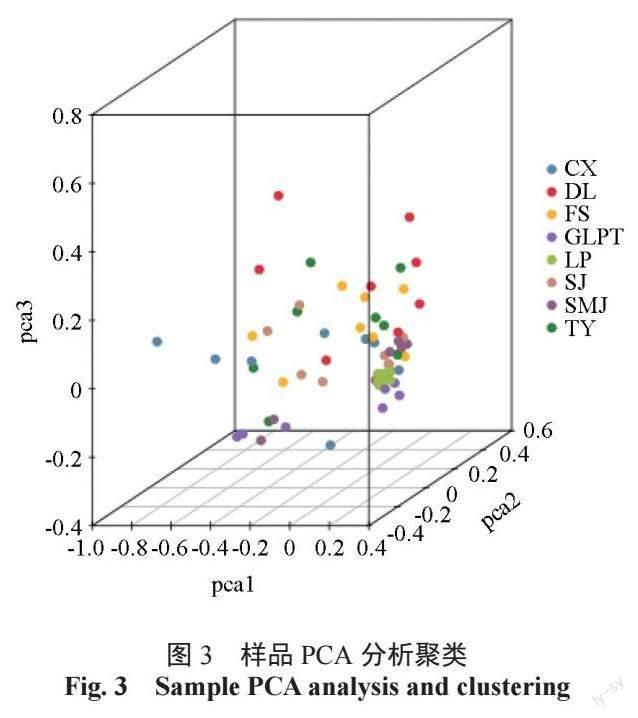
主成分分析(PCA,principal component analysis)是一种利用线性代数方法,把多个变量变为少数几个综合变量数据,相对全面地体现出整个数据量特征,进行由繁到简的降维研究方法。我们把这些所谓的综合变量称为主成分,几个主成分之间彼此不相关,也就说明他们之间含有不重复的信息。基于以上筛选获得的SNP,通过cluster 软件,分别设置3 个变量为主成分代表,对所有64份供试油茶种质进行主成分分析,由此得到一个三维的聚类图(图3)。结果显示广西凤山中籽茶居群内的个体相对集中于一个层面,居群间存在一定程度的群体分层,部分居群间个体少量互有混杂,暗示广西凤山县境内的不同居群的中籽茶可能由于距离、时间、小气候等因素产生的一定差异,同时也一定程度上保留部分共有基因、性状。PCA 分析与遗传进化聚类结果较一致。
3 讨 论
SLAF-seq 技术是一种基于高通量测序技术,在没有供试物种全基因组序列信息的前提下,通过对供试物种一定数量的个体进行简化基因组测序,从而获取该物种基因组中相对多的特异DNA 序列,用以开发大量的特异SNP 标记,并可与前期的重要农艺性状关联分析,以实现候选功能区域的精细定位。SLAF-seq 技术因具有高通量、高效率、高准确率、成本低等优势,已被广泛应用在小麦、棉花、白菜、黄瓜等多个物种的研究中[14-16]。
相对于RAPD、SRAP、ISSR、SSR 等第一、二代分子标记均是依赖于电泳检测的分析技术,PCR 扩增过程以及条带读取受人为因素以及环境的影响较大,高通量样本分析工作量大,而且这些标记在基因组中呈现不均匀分布,油茶为异花授粉植物,不同种质的杂合度高,遗传背景复杂,已开发的油茶分子标记数量极为有限,因此,难以全面、准确地反映油茶群体的遗传多样性,这也限制了优良油茶种质资源的开发和利用。
酶切是直接影响后续测序及分析关键指标,故SLAF-seq 测序前应先进行酶切预测,先酶切预测供试物种近缘物种基因组,其原理是借用酶切参考基组与供试物种实际参考基因组的大小比例对开发得到的标签数目进行换算。油茶为异花授粉植物、杂合度高、遗传背景复杂,目前尚无可参考基因组,所以根据预测的普通油茶基因组大小以及GC 含量等信息(实际基因组大小约为2.94Gb,GC 含量为44.85%),最终选取茶树(组装出的基因组大小为3.1 Gb,GC 含量为42.30%)基因组作为酶切参考基因组;为评估酶切实验的准确性,选用水稻作进行测序对照,结果显示水稻双端比对效率为95.53%,酶切成功效率为95.06%,表明SLAF 建库正常,表明选用茶树基因组作为参考基因组进行酶切预测是可行的。SLAF-seq 自开发以来已广泛地应用于动植物高密度遺传连锁图谱构建、QTL 定位、群体遗传学分析等研究中并取得了良好的效果[17-21]。尽管前人开发了大量的DNA 分子标记对油茶群体种质资源进行遗传多样性及差异化分析,包括RAPD、SRAP、ISSR、SSR 等分子标记[22-27]。但由于第一、二代分子标记均是使用电泳检测分离技术,辨别度和灵敏度不高,大量样本分析比较繁杂,而且这些标记在基因组中的分布不均匀,难以全面、准确地反映油茶群体的遗传多样性。
广西凤山中籽茶原始野生种质资源长期未得到有效保护,并缺乏有效经营管护措施,加之居群内植株树龄偏大(50 ~ 60 a)、且前期植株密度过大造成树体畸形、老化严重,导致其原始自然分布居群数较少、居群内正常发育单株极少,造成本研究供试材料的居群内植株选取的基数偏少(每个居群内8 株)。但在有限的供试材料选取采集过程中,严格把控选取供试材料质量(如:植株间距离、树龄相对一致、无病虫害、叶片完整度、种实数量等因素),最大限度地体现广西凤山中籽茶种质资源遗传分化深度及广度。本研究基于SLAF-seq 技术对广西凤山中籽茶8 个居群64 份种质进行SNP 标记开发及差异化分析,获得的有效标记信息丰富,远超过以往研究油茶种质标记开发数量总和。因此,油茶全基因组范围内的分子标记开发与利用显得尤为重要。
4 结 论
本研究首次从DNA 水平上对广西凤山中籽茶开展了遗传多样性研究工作,并获得了199 745 个高一致性的SNP 标记位点,进一步表明SLAF-seq测序技术可有效用于油茶群体种质SNP标记开发,且标记数量、质量及效率远远优于ISSR、SRAP、SSR 等第一、二代分子标记技术。凤山中籽茶群体遗传聚类分析表明,广西凤山县境内的不同居群的中籽茶可能由于距离、时间、小气候等因素产生的一定差异,同时也一定程度上保留部分共有基因、性状;其遗传变异多来自于居群内,居群间各群各自聚类、区域性相对明显,其遗传分化程度与地理距离远近呈现正相关性。本研究基于简化基因组技术开发出的大量凤山中籽茶群体SNP 标记,可为下一步油茶种质优异性状QTL 定位及辅助育种奠定基础,为广西其他特色地方品种的相关研究提供有益的参考。
參考文献:
[1] 胡芳名, 谭晓风, 裴东等. 我国经济林学科发展报告[C]// 国家林业局, 广西壮族自治区人民政府, 中国林学会. 第二届中国林业学术大会—S9 木本粮油产业化论文集.[ 出版者不详],2009:14.
HU F M, TAN X F, PEI D, et al. Report on the developmentof China's economic forestry discipline [C]//The StateForestry Administration, the People's Government ofGuangxi, the Chinese Forestry Society. The 2nd China forestryacademic conference-S9 woody grain and oil industrializationanthology [Publisher unknown], 2009:14.
[2] 庄瑞林. 我国油茶良种选育工作的历史回顾与展望[J]. 林业科技开发,2010,24(6):1-5.
ZHUANG R L. Historical review and prospect on the selectionof improved oil tea cultivars in China[J]. China Forestry Scienceand Technoloy,2010,24(6):1-5.
[3] 李良. 湖北省油茶优良单株遗传多样性及品质初步分析[D].武汉: 华中农业大学,2010.
LI L. Analysis of genetic diversity and quality ofCamellia oleiferaresources in Hubei province[D]. Wuhan: Huazhong AgriculturalUniversity,2010.
[4] 戴瑞强, 张林, 扈东青, 等. 植物分子育种研究进展[J]. 安徽农业科学,2009,37(32):15725-15727, 15729.
DAI R Q, ZHANG L, HU D Q, et al. Research advance in plantmolecular breeding[J]. Journal of Anhui Agricultural Science,2009,37(32):15725-15727,15729.
[5] 傅志强, 袁汕, 申春晖等. 基于花器官特征的油茶遗传多样性分析[J]. 经济林研究,2022,40(4):12-18,28.
FU Z Q, YUAN S, SHEN C H, et al. Genetic diversity analysisbased on characteristics of ?oral organs ofCamellia oleifera[J].Non-wood Forest Research,2022,40(4):12-18,28.
[6] 王滑, 郝俊民, 王宝庆, 等. 中国核桃8 个天然居群遗传多样性分析[J]. 林业科学,2007,43(7):120-124.
WANG H, HAO J M, WANG B Q, et al. SSR analyses of geneticdiversity of eight natural walnut populations in China[J]. ScientiaSilvae Sinicae,2007,43(7):120-124.
[7] 周炎花, 乔小燕, 马春雷, 等. 广西茶树地方品种遗传多样性和遗传结构的EST-SSR 分析[J]. 林业科学,2011,47(3):59-67.
ZHOU Y H, QIAO X Y, MA C L, et al. Genetic diversity andstructure of tea landraces from Guangxi based on EST-SSRanalysis[J]. Scientia Silvae Sinicae,2011,47(3):59-67.
[8] 刘凯, 王东雪, 江泽鹏, 等. 基于SLAF-seq 技术的油茶SNP 位点开发及杂交后代早期鉴定[J]. 广西林业科学,2018,47(1):13-17.
LIU K, WANG D X, JIANG Z P, et al. SNP markers developmentbased on SLAF-seq and identification ofCamellia Oleifera Abelhybrids[J]. Guangxi Forestry Science,2018,47(1):13-17.
[9] SUN X W, LIU D Y, ZHANG X F, et al. SLAF-seq: an efficientmethod of large-scalede novo SNP discovery and genotypingusing high-throughput sequencing[J]. PLoS One,2013,8(3):236078820.
[10] LI R Q, YU C, LI Y R, et al. SOAP2: an improved ultrafasttool for short read alignment[J]. Bioinformatics,2009,25(15):1966-1967.
[11] LI H, DURBIIN R. Fast and accurate short read alignment withBurrows-Wheeler transform[J]. Bioinformatics,2009,25(14):1754-1760.
[12] MCKENNA A, HANNA M, BANKS E, et al. The genomeanalysis toolkit: a MapReduce framework for analyzing nextgenerationDNA sequencing data[J]. Genome Research,2010,20(9):1297-1303.
[13] LI H, HANDSAKER B, WYSOKER A, et al. The sequencealignment/map format and SAMtools[J]. Bioinformatics,2009,25(16):2078-2079.
[14] MINGJIAN H, HAIPING Z, KAI L, et al. Cloning andcharacterization ofTaTGW-7A gene associated with grain weightin wheat via SLAF-seq-BSA[J]. Frontiers in Plant Science,2016,7:1-12.
[15] ZHANG Z, SHANG H H, SHI Y Z, et al. Construction of ahigh-density genetic map by specific locus amplified fragmentsequencing (SLAF-seq) and its application to Quantitative TraitLoci (QTL) analysis for boll weight in upland cotton (Gossypiumhirsutum.)[J]. BMC plant biology,2016,16(1):79.
[16] LIANG D N, CHEN M Y, et al. QTL mapping by SLAF-seq andexpression analysis of candidate genes for aphid resistance incucumber[J]. Frontiers in Plant Science,2016,7(7):1-8.
[17] LI B, TIAN L, ZHANG J, et al. Construction of a high-densitygenetic map based on large-scale markers developed by specificlength amplified fragment sequencing (SLAF-seq) and itsapplication to QTL analysis for isoflavone content in glycinemax[J]. BMC Genomics,2014,15:1086.
[18] XU X W, XU R X, ZHU B Y, et al. A high-density genetic mapof cucumber derived from specific length amplified fragmentsequencing (SLAF-seq)[J]. Frontiers in Plant Science,2014,5:768.
[19] XU X W, LU L, ZHU B Y, et al. QTL mapping of cucumber fruitflesh thickness by SLAF-seq[J]. Scientific Reports,2015,5:15829.
[20] GENG X X, JIANG C H, YANG J, et al. Rapid identification ofcandidate genes for seed weight using the SLAF-Seq method inBrassica napus[J]. PLoS One,2016,11(1):e147580.
[21] 俞超, 陳煜, 汪财生, 等. 基于SLAF-seq 技术的红心火龙果SNP 位点开发及遗传分析[J]. 热带作物学报,2017,38(4):591-596.
YU C, CHEN Y, WANG C S, et al. SNP sites by specific lengthamplification fragment sequencing (SLAF-seq) and geneticanalysis in red pitaya[J]. Journal of Tropical Crops,2017,38(4):591-596.
[22] 王保明, 陈永忠, 谭晓风, 等. 应用ISSR 分析油茶无性系的遗传多样性[J]. 东北林业大学学报,2008,36(6):19-23.
WANG B M, CHEN Y Z, TAN X F, et al. Genetic diversity ofelite clones ofCamellia oleifera by ISSR[J]. Journal of NortheatForestry University,2008,36(6):19-23.
[23] 黄永芳, 陈锡沐, 庄雪影, 等. 油茶种质资源遗传多样性分析[J]. 林业科学,2006,42(4):38-43.
HUANG Y F, CHEN X L, ZHUANG X Y, et al. 2006, Analysisof genetic diversity inCamellia oleifera germplasms[J]. ScientiaSilvae Sinicae,2006,42(4):38-43.
[24] 林萍, 姚小华, 王开良, 等. 油茶长林系列优良无性系的SRAP分子鉴别及遗传分析[J]. 农业生物技术学报,2010,18(2):272-279.
LIN P, YAO X H, WANG K L, et al. Identification and geneticanalysis of Changlin series superior clones by SRAP molecularmarker[J]. Journal of Agricultural Biotechnology,2010,18(2):272-279.
[25] 梁国校, 刘凯, 马锦林, 等. 香花油茶分子分类与鉴定[J]. 经济林研究,2017,35(1):26-29.
LIANG G X, LIU K, MA J L, et al. Molecular taxonomy andidentification ofCamellia osmantha[J]. Non-wood Forest Research,2017,35(1):26-29.
[26] 謝荟, 祝文娟, 张应中, 等. 基于SRAP 分子标记的广宁红花油茶种质资源遗传多样性研究[J]. 中南林业科技大学学报,2017,37(8):93-97.
XIE H, ZHU W J, ZHANG Y Z, et al. Genetic diversity ofCamellia semiserrata Chi germplasm based on SRAP markers[J].Journal of Central South University of Forestry & Technology,2017,37(8):93-97.
[27] 高园, 解元, 杨自云, 等. 基于花期差异和ISSR-PCR 分析的不同茶梅品种间亲缘关系[J]. 经济林研究,2022,40(4):209-217.
GAO Y, JIE Y, YANG Z Y, et al. Genetic relationship analysis ofdifferentCamellia sasanqua cultivars based on the difference of?owering season and ISSR-PCR[J]. Non-wood Forest Research,2022,40(4):209-217.
[ 本文编校:李义华]
猜你喜欢
音乐教育与创作(2019年4期)2019-11-14
现代园艺(2017年22期)2018-01-19
现代园艺(2017年22期)2018-01-19
现代园艺(2017年13期)2018-01-19
现代园艺(2017年21期)2018-01-03
现代园艺(2017年21期)2018-01-03
中国西部(2017年4期)2017-04-26
湖南农业(2017年1期)2017-03-20
源流(2017年1期)2017-03-07
广西林业科学(2016年2期)2016-03-20
