
CHO细胞外源抗体基因拷贝数实时定量PCR检测方法的建立及验证
2024-01-26 12:17张慧高文丽李凤智胡加亮李剑
中国生物制品学杂志 2024年1期
张慧,高文丽,李凤智,胡加亮,李剑
1.中国药科大学生命科学与技术学院,江苏 南京 211100;2.天士力生物医药股份有限公司,上海 201203
随着细胞工程的不断发展,越来越多的生物表达系统可用于大规模生产蛋白,其中原核和真核表达系统是最主要的两类。而真核表达系统中的哺乳动物细胞表达系统由于具有翻译后修饰的能力,能够保证蛋白的天然活性,在生物制药领域发挥着重要作用,目前已成为表达和生产部分蛋白药物、基因工程抗体等目的蛋白的首选方式[1-4]。目前市面上超过50%的蛋白类药物是使用哺乳动物细胞生产的,这些蛋白类药物在分子结构和生物化学性质方面与人类天然蛋白具有相似性[5]。经重组表达载体构建、转染、筛选和鉴定等一系列步骤,获得稳定表达外源蛋白产物的细胞株尤为重要。外源基因转染至宿主细胞后,随机插入宿主细胞基因组DNA 上,插入的拷贝数并不固定,需检测外源基因拷贝数和基因表达水平[5],以检验细胞株是否具有传代稳定性。
Southern blot 是检测基因拷贝数的传统方法,荧光原位杂交(fluorescencein situhybridization,FISH)、基因组杂交等分子杂交方法也可用于检测基因拷贝数变化,但这些方法均存在局限性[6]。Southern blot费时费力,同时由于无DNA扩增过程,需大量的高质量基因组DNA样品。1995年,美国Perkin Elmer公司开发的荧光定量PCR技术问世,成为一种新的定量技术[7],相较于Southern blot 等方法,实时定量PCR 法操作简便,DNA样品用量少,可快速且准确地检测基因拷贝数,该方法可对反应过程中每一循环的PCR产物积累进行检测,而不是通过常规PCR 方法进行终点检测,对于DNA 产物的积累可通过多种荧光化学方法来检测[8-9],如可与所有双链DNA 结合的染料SYBR GreenⅠ和特异性结合的探针TaqMan[10-11],本实验选择了更经济的SYBR GreenⅠ染料。
SYBR GreenⅠ染料能够结合于所有双链DNA双螺旋小沟区域,在激发光照射下产生荧光,所产生的荧光强度与PCR 产物的量有关,根据Ct值与起始模板数的相关性方程,将样品的Ct值代入即可获得目的基因的起始模板数[12-13]。虽然所有模板均被稀释至同一浓度,但基因组样品质量不同、基因组大小不同以及存在取样误差等原因,导致定量分析反应体系中模板的浓度略有差异,因此需引入内参基因来标准化样品,以校正样品因初始浓度不同而造成的差异[14-15]。由于B2m(β2-microglobulin)基因可在CHO 细胞系中具有稳定的表达水平[16-17],选择该基因作为内参基因,检测外源导入的轻链(light chain,LC)和重链(heavy chain,HC)基因相对于B2m基因的拷贝数。本研究旨在采用SYBR GreenⅠ染料建立针对CHO 细胞株LC和HC基因拷贝数的实时定量PCR检测方法,并对建立的方法进行验证及初步应用。
1 材料与方法
1.1 质粒及细胞 含外源LC、HC基因和内参基因的1∶1标准质粒pMV-1962-LCHC-B2m(大小5 104 bp)由深圳华大基因股份有限公司合成并提供;未转入表达质粒的阴性对照CHO 细胞和转染抗体基因的CHO细胞株均由天士力生物医药股份有限公司研发部提供。
1.2 主要试剂及仪器 基因组DNA抽提试剂盒(TaKaRa MiniBEST Universal Genomic DNA Extraction Kit Ver.5.0)、蛋白酶K 和RNA 酶均购自日本TaKaRa 公司;PowerUp SYBRTMGreen Master Mix 染料预混液和100 bp DNA marker购自美国Thermo公司;纯水Nuclease-free Water 购自北京全式金生物技术股份有限公司;Light Cycle 480Ⅱ荧光定量PCR仪购自瑞士Roche公司;超微量分光光度计购自德国IMPLEN公司。
1.3 引物设计及合成 利用Primer Premier 5 软件设计LC、HC基因的特异性引物,由深圳华大基因股份有限公司合成,用纯水溶解至10 µmol/L。引物序列见表1。
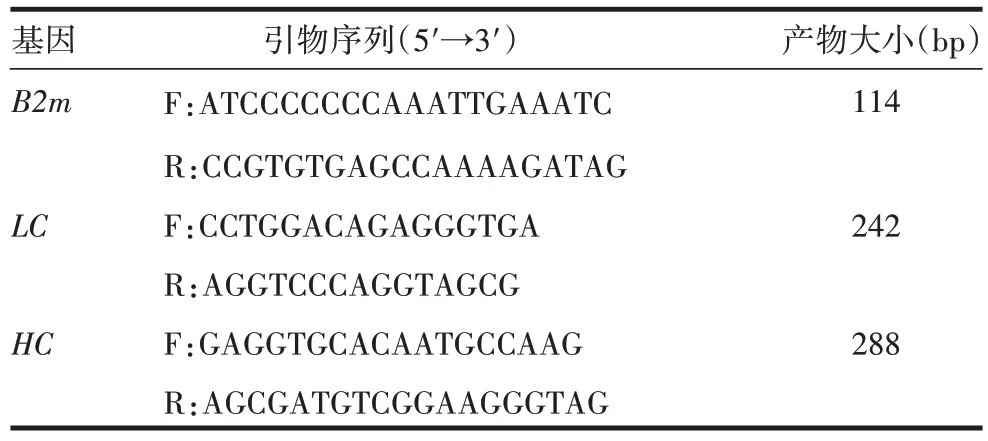
表1 实时定量PCR所用引物序列与产物大小Tab.1 Primer sequences and product size of real-time quantitative PCR
1.4 样品制备 按照TaKaRa MiniBEST Universal Genomic DNA Extraction Kit Ver.5.0 试剂盒说明书,分别提取阴性对照CHO 细胞和表达抗体的工作细胞库(WCB)细胞的基因组DNA,溶于纯水中,用紫外分光光度计检测DNA 浓度。用纯水将基因组DNA稀释至10 ng/µL,作为定量PCR检测的阴性对照和样品,以标准质粒作为阳性对照,纯水作为空白对照。
1.5 实时定量PCR反应体系及反应程序 反应体系共20 µL:PowerUp SYBRTMGreen Master Mix 10 µL,正、反向引物(10µmoL/L)各0.2µL,DNA模板1µL,纯水8.6µL。反应条件:95 ℃30 s,95 ℃10 s,60 ℃30 s,72 ℃5 s,共40个循环。熔解曲线设置:95 ℃5 min,65 ℃1 min。当温度上升,定量PCR产物逐渐解链形成单链DNA,SYBR GreenⅠ染料结合的双链DNA 减少,在一个温度区间内,荧光信号急速下降,对荧光信号下降速率进行分析,若形成单一峰,表明管内产物一致且无污染。
1.6 方法的验证
1.6.1 特异性 以阳性对照、阴性对照、WCB 细胞基因组DNA 和空白对照为模板,查看熔解曲线是否呈单一峰[18],并收集WCB 细胞基因组DNA 扩增产物,经1%琼脂糖凝胶电泳鉴定。
1.6.2 线性 采用SYBR GreenⅠ荧光相对定量PCR法,使用两种引物,同时对目的基因LC、HC和内参基因B2m进行定量PCR,通过ΔΔCt法计算目的基因相对倍数[19]。该方法要求目的基因和内参基因引物具有尽可能相近的扩增效率。根据实时荧光定量PCR国际化标准——MIQE 指南,两对引物的扩增效率应在90%~110%之间,且扩增效率差异小于10%。
按照下式计算拷贝数,将标准质粒10 倍系列稀释成5×106、5×105、5×104、5×103拷贝/µL,获得线性方程,根据斜率(k)与扩增效率(E)之间的关系,对引物扩增效率进行验证。
拷贝数(copies/µL)=(6.02×1023)×(ng/µL×10-9)/(DNA length×660);
E(%)=(10-1/k-1)×100%
1.6.3 精密性 重复性:取10 ng/µL 的WCB 细胞基因组DNA,由同一名实验人员在同一次试验中测定9 个独立平行供试品的Ct值,计算相对标准偏差(relative standard deviation,RSD)。中间精密度:由另1名实验人员在2个时间点测定同一样品9份独立平行供试品的Ct值,计算18个测得值的RSD。
1.6.4 耐用性 将WCB 细胞基因组DNA 置-20 ℃冻融1和5次后进行定量PCR 检测,测定结果与未经冻融的WCB 细胞基因组DNA 的Ct值进行比较,计算差异百分比(relative percent difference,RPD)。
1.7 方法的初步应用 按照TaKaRa MiniBEST Universal Genomic DNA Extraction Kit Ver.5.0 试剂盒说明书,分别提取WCB、细胞群倍加数(population doubling level,PDL)10、PDL20、PDL30、PDL40、PDL50、PDL60、PDL70、PDL80 的细胞基因组DNA(PDL 是经体外培养后细胞数翻倍的次数,在二倍体细胞中,细胞代次基本等同于细胞数双倍扩增次数)。用建立的荧光定量PCR 检测方法对稀释至10 ng/µL 的样品重复检测3 次,采用ΔΔCt法,将传代细胞拷贝数与WCB 细胞拷贝数进行比较,所得结果即为该细胞株拷贝数稳定情况。
1.8 数据采集及分析 应用RocheLightCycle480software release 1.5.0 SP4 软件处理数据,Graphpad prism 8.0.1软件绘图。
2 结果
2.1 方法的验证
2.1.1 特异性 样品和阳性对照的3 对引物熔解曲线呈单一特征峰,阴性对照中加入B2m引物其熔解曲线也呈单一峰。以不同DNA模板对不同基因进行扩增,结果均符合预期,其中样品扩增产物均显示单一条带,大小符合预期,表明样品中不存在非特异性扩增,引物可特异性结合模板DNA。见图1和图2。
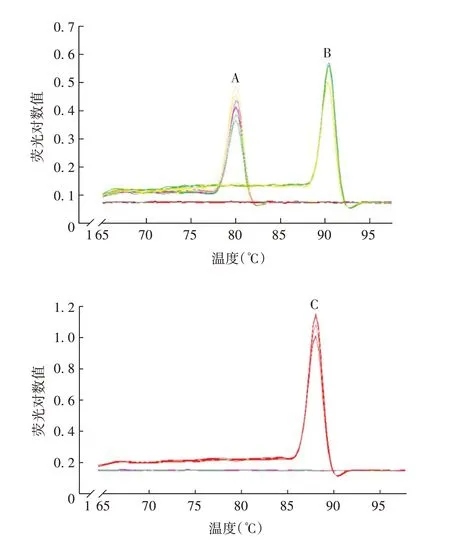
图1 B2m、LC、HC基因的熔解曲线Fig.1 Melt curves of genes B2m,LC and HC

图2 B2m、LC、HC基因扩增产物电泳图Fig.2 Electrophoretic profile of amplification products of genes B2m,LC and HC
2.1.2 线性 3对引物的扩增曲线和标准曲线见图3 ~5。B2m、LC、HC线性方程分别为:y=-3.17 lgx+41.17、y=-3.18 lgx+41.13、y=-3.35 lgx+39.48,相关系数(R2)分别为0.996 1、0.995 9、0.999 2,根据斜率计算出3对引物的扩增效率分别为106.7%、106.3%、99.1%,均接近100%,且LC和HC引物的扩增效率与B2m相差小于10%。选用这些引物进行反应,其扩增产物可真实反映模板拷贝数。

图3 内参基因B2m的扩增曲线(A)和标准曲线(B)Fig.3 Amplification curve(A)and standard curve(B)of reference gene B2m
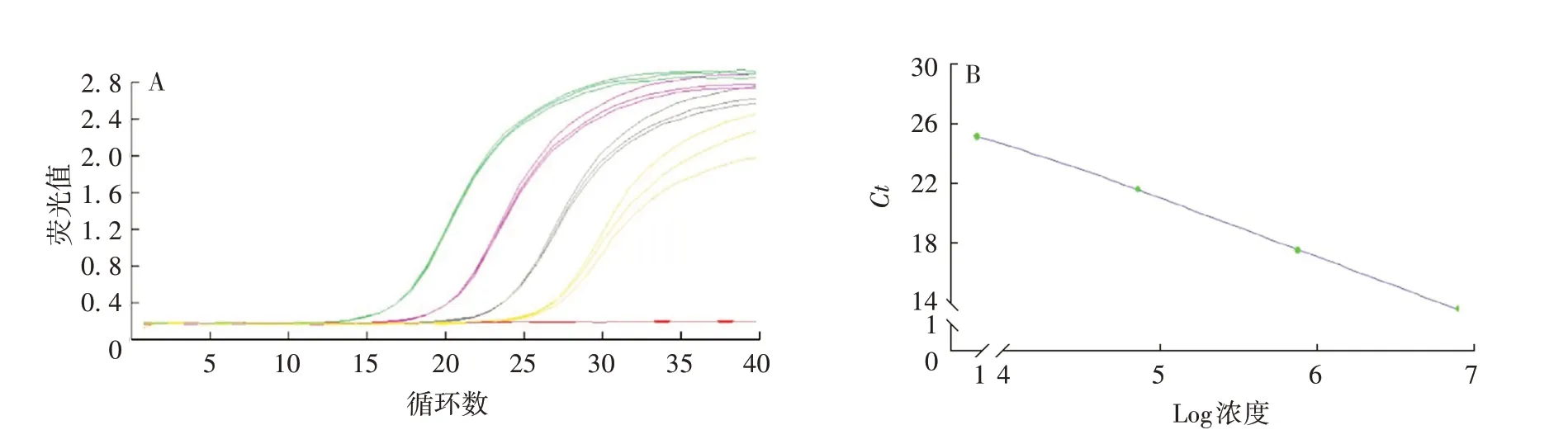
图4 外源基因LC的扩增曲线(A)和标准曲线(B)Fig.4 Amplification curve(A)and standard curve(B)of exogenous gene LC
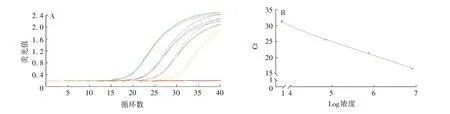
图5 外源基因HC的扩增曲线(A)和标准曲线(B)Fig.5 Amplification curve(A)and standard curve(B)of exogenous gene HC
2.1.3 精密性
2.1.3.1 重复性 同一名实验人员在同一次试验中重复测定9 份平行供试品的Ct值差异较小,RSD均小于1%,见表2。表明该方法重复性良好。
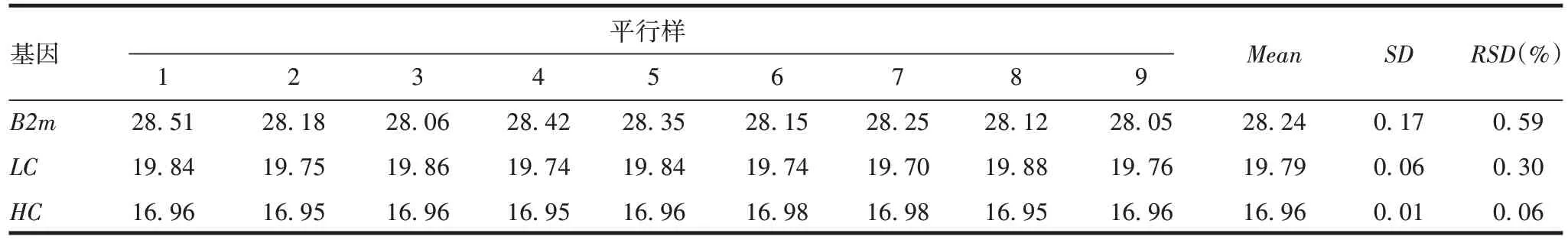
表2 方法的重复性验证结果(Ct)Tab.2 Reproducibility verification results(Ct)
2.1.3.2 中间精密度 另一名实验人员在不同日期测定9 份独立平行供试品Ct值的RSD均小于1%,见表3。表明该方法具有良好的重复性和准确性。
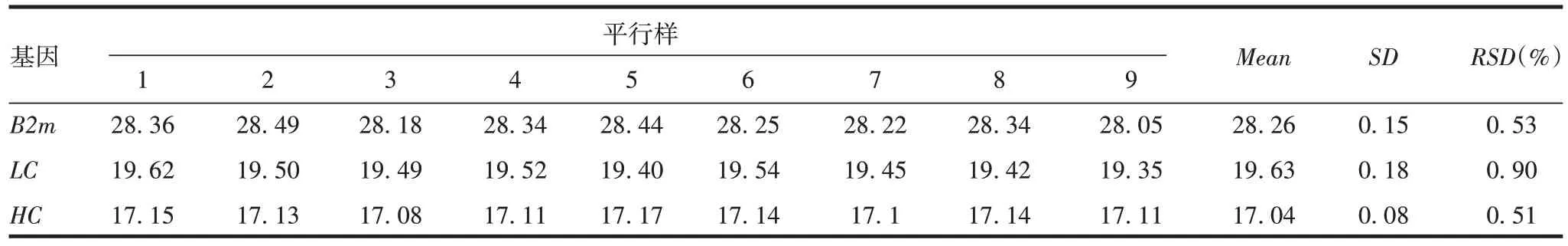
表3 方法的中间精密性验证结果(Ct)Tab.3 Intermediate precision verification results(Ct)
2.1.4 耐用性 WCB 细胞基因组DNA 置-20 ℃分别冻融1 和5 次的RPD 均小于1%,见表4。表明少次冻融对样品影响较小。
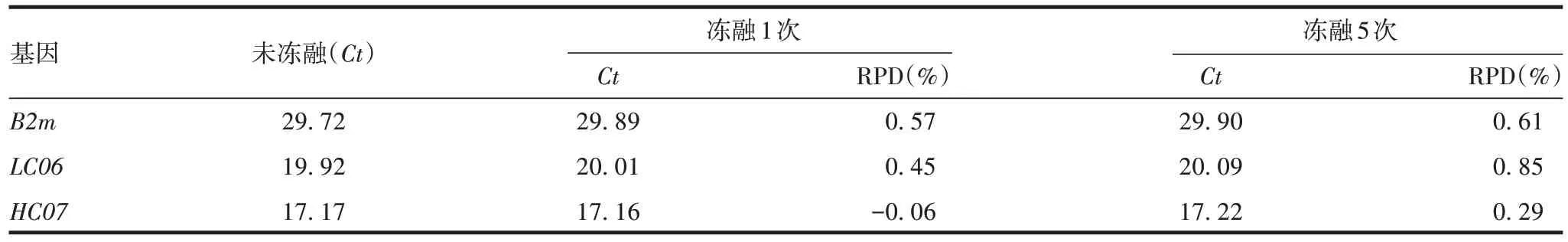
表4 方法的耐用性验证结果Tab.4 Durability verification results
2.2 方法的初步应用 结果可见,样品PCR 扩增反应良好,同一引物的样品其熔解曲线呈单一峰,PCR扩增的特异性良好,且随着细胞传代次数的增加,LC、HC基因拷贝数均未见明显变化。见表5和表6。
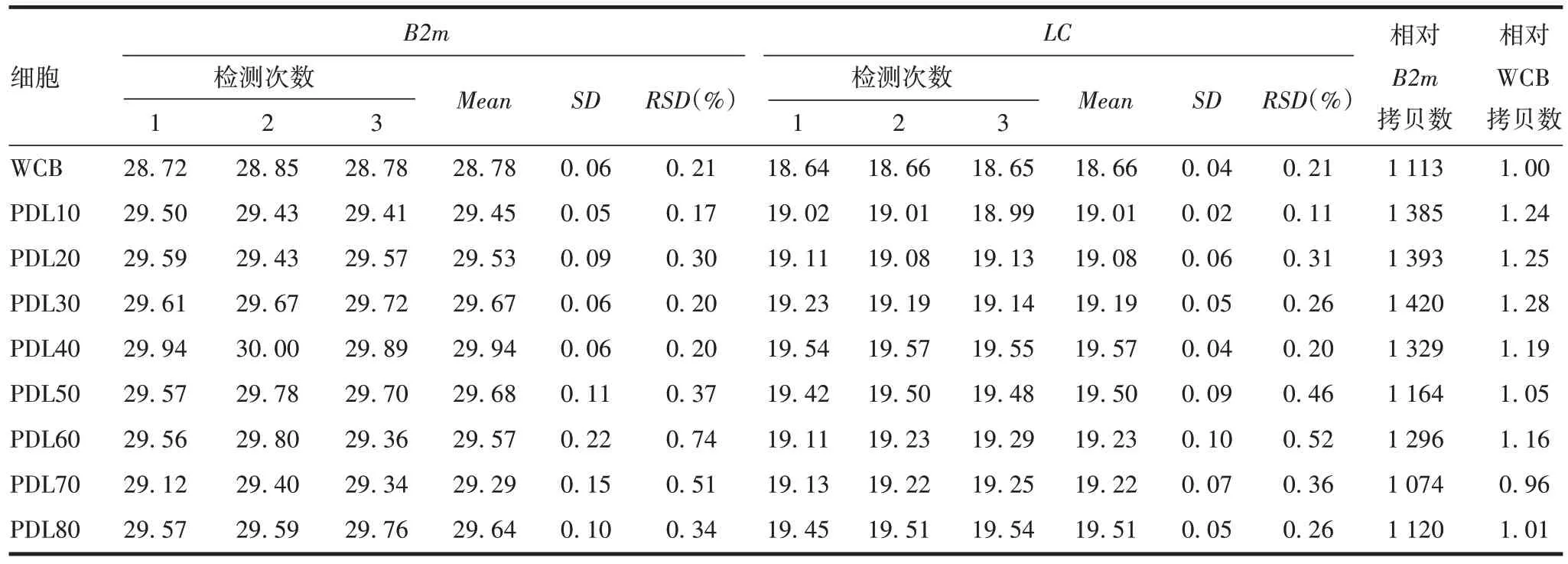
表5 不同代次CHO细胞中LC基因拷贝数Tab.5 LC gene copy numbers in different generations of CHO cells
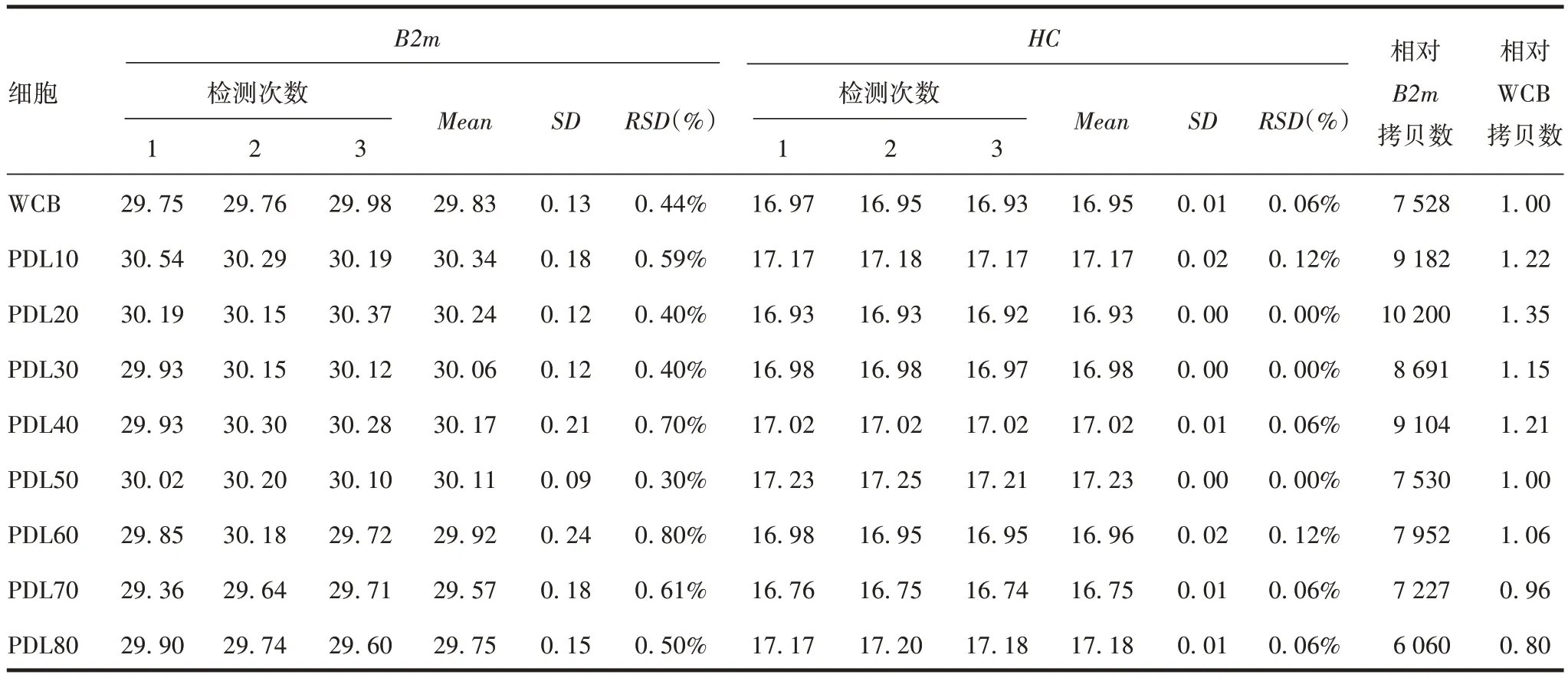
表6 不同代次CHO细胞中HC基因拷贝数Tab.6 HC gene copy numbers in different generations of CHO cells
3 讨论
表达外源蛋白的重组细胞株的稳定性对于产品质量的稳定具有关键意义。转染外源基因的CHO细胞在连续升高的叶酸类似物氨甲蝶呤(amethopterin,MTX)作用下加压扩增外源基因,使得抗体LC、HC在CHO 细胞基因组中拷贝数不断增加[20]。为了确保这些基因能够稳定存在于基因组中,需测定不同代次细胞的拷贝数,并进行比较。常用的实时定量PCR 方法有Taqman 探针法和SYBR GreenⅠ染料法,均可用于拷贝数的测定。Taqman 探针法除需设计引物外,还需设计与模板特异性结合的探针,相较于SYBR GreenⅠ染料法,其耗时长,成本高[21-22]。本研究选择了SYBR GreenⅠ染料法,其成本低,但由于该染料可与任意双链DNA 结合,包括引物二聚体和非特异性产物,因此需通过熔解曲线和琼脂糖凝胶电泳共同判断其产物是否单一无污染,以及产物大小是否符合预期,以验证引物的特异性[23-25]。
对引物的性能进行确证是建立此方法的关键。对目的基因与内参基因拷贝数为1∶1 的标准质粒进行梯度稀释,制备10 倍浓度梯度的标准模板,用这些模板进行扩增并建立了标准曲线。3 对引物的标准曲线R2均大于0.99,扩增效率相差不超过8%,且均在95%~110%之间,表明引物与模板DNA能够极大程度地互补,且几乎无非特异性扩增。引物设计保障了方法的准确性,扩增产生的Ct值差异能够真实地比较出不同模板间拷贝数的差异。
有研究表明,单个样品扩增反应的Ct值小于25时,其SD小于0.3即认为结果可信,能够准确反映样品的拷贝数[26]。而本方法精密性验证试验中,Ct值的SD均小于0.3,RSD小于1%,符合该标准,表明该方法准确可靠,重复性良好。
模板DNA 的保存是保证DNA 质量的重要环节。低温冷冻保存是模板DNA 普遍的保存方式,但在反复冻融过程中,其物理结构易受破坏,不利于后续试验。在耐用性验证试验中,反复5 次冻融对DNA 影响较小,为模板DNA使用次数提供了参考。
采用建立的实时定量PCR 法检测各传代细胞,发现细胞株在传代至PDL80 内与WCB 比较,LC、HC基因拷贝数均未见显著变化,表明该重组细胞株外源基因拷贝数具有较好的传代稳定性。
综上所述,本研究成功建立了实时定量PCR 法检测重组CHO 细胞外源基因拷贝数,可用于研究外源基因在CHO 细胞中的遗传稳定性,该方法简便快速,结果可靠,为后续CHO 细胞株表达的外源基因拷贝数检测提供了参考。
猜你喜欢
舰船科学技术(2022年11期)2022-07-15
河北医学(2021年10期)2021-10-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
西藏农业科技(2019年3期)2019-11-04
中国临床医学影像杂志(2019年6期)2019-08-27
中成药(2018年12期)2018-12-29
现代园艺(2018年3期)2018-02-10
中成药(2017年6期)2017-06-13
上海农业学报(2017年3期)2017-04-10
医学研究杂志(2015年4期)2015-06-10
