
CRISPR/Cas9核糖核蛋白介导的无T-DNA插入的白桦BpGLK1精准突变
2024-01-23 05:45邱志楠白向东刘桂丰
南京林业大学学报(自然科学版) 2024年1期
王 伟, 邱志楠, 李 爽, 白向东,刘桂丰,姜 静
(林木遗传育种全国重点实验室(东北林业大学),黑龙江 哈尔滨 150040)
CRISPR/Cas9技术是继锌指核酸酶(zinc-finger nucleases,ZFNs)、TALEN核酸酶(transcription activator like effector nucleases,TALENs)之后的第3代基因编辑技术[1-2],该技术与ZFNs/TALENs技术比较,其操作方法简便,敲除基因效率更高,编辑更精准,大大降低了编辑脱靶机率。目前已成功地在拟南芥(Arabidopsisthaliana)、烟草(Nicotianabenthamiana)、棉花(Gossypiumhirsutum)、水稻(Oryzasativa)、玉米(Zeamays)、大豆(Glycinemax)、苹果品种(Malusprunifolia‘Seishi’ ×M.pumilavar.paradisiaca‘M.9’)和葡萄(Vitisvinifera)等植物基因组定点编辑研究中广泛应用。大量研究表明该技术是作物品质改良、提高产量及抗逆性的强大工具[3-7]。通常CRISPR/Cas9技术主要采用根癌农杆菌介导法在T-DNA的驱动下,将Cas9蛋白-gRNA-核糖核蛋白以及选择标记基因导入受体细胞,进而通过选择培养将非转化细胞杀死,而转化细胞由于获得标记基因的抗性而成活下来,最终获得基因组编辑植物。此类方法在获得基因组编辑的植物基因组中仍含有插入的T-DNA(包括载体及标记基因)序列等[3],筛选标记基因虽为转化植株的筛选提供了方便,但一旦完成筛选得到所需要的转化植株之后,选择标记基因就变成多余的,甚至有潜在危害[8]。近年来,科研人员开发了一种基于CRISPR/Cas9核糖核蛋白(RNP)的无外源基因整合的基因编辑系统,即在体外将Cas9蛋白与sgRNA进行预组装形成复合物,然后通过微弹轰击转化方式直接导入受体细胞核中,进而完成对靶基因的定点突变[9]。该技术的主要特点是避免了外源基因及选择标记转基因插入的干扰。目前,已在拟南芥、烟草和水稻等植物中实现了无T-DNA插入的定向基因编辑,获得的基因编辑突变体与天然存在的遗传变异毫无区别[10]。因此,探究无选择标记转基因技术研究已成为当今的热点之一。
确定适合标记靶基因是探究无选择标记转基因技术研究的第一步。广泛存在于植物中的GLK转录因子属于GARP超家族中的一员,在调控叶绿体发育等方面起着十分关键的作用[11]。该转录因子参与调控核定位的叶绿体蛋白以及与光合作用相关基因的表达[12],研究证明,白桦glk缺失突变株、白桦BpGLK1干扰表达及银中杨PaGLK1干扰表达株系的叶色均为鲜艳的黄绿色[13-14],不仅表明GLKs基因在调控植物叶绿素合成进而影响叶色方面至关重要,同时也可以BpGLK1作为靶基因,其功能失活后可提供一个易于肉眼观察的明显叶色变化。因此,本研究以白桦为受体,利用CRISPR/Cas9核糖核蛋白(RNP)的无外源基因整合的基因编辑系统,进行BpGLK1特定靶点的定向编辑,研究结果可为创制林木无T-DNA插入突变体提供崭新思路。
1 材料与方法
1.1 试验材料
白桦(Betulaplatyphylla×B.pendula)种子采自东北林业大学白桦强化种子园内自由授粉的优树,将采收的种子晾干,去除苞片等杂质后,塑料袋塑封后置于-20℃冰箱保存备用。
PCR纯化试剂盒(QIAGEN 28206),胶回收试剂盒(QIAGEN 28706),T7快速高产RNA合成试剂盒(NEB E2050S),BstEII(NEB R0162V),Cas9蛋白(质量浓度为1 mg/mL)由林木遗传育种全国重点实验室(东北林业大学)姜立泉教授团队惠赠,pAbAi质粒(4 894 bp)由东北林业大学林木遗传育种研究团队保存,金粒(Biorad)。
1.2 试验方法
1.2.1BpGLK1第1外显子的克隆及重组载体的构建
根据白桦BpGLK1(Bpev01.c0167.g0013.m0001)基因序列,在BpGLK1第1外显子BpGLK1-E1(946 bp)序列前后设计上下游引物BpGLK1-E1-F.5′-CGGGGTACCGCTCACGAGCTGCCACT-3′;BpGLK1-E1-R.5′-ACGCGTCGACAGCTGCTCCACAGCTTGC-3′。以白桦DNA为模板进行PCR扩增,获得BpGLK-E1片段。用KpnI和SalI分别双酶切pAbAi质粒及BpGLK1-E1片段,酶切产物用T4连接酶连接后并转化大肠杆菌,挑取阳性克隆进行菌液PCR检测,并送往擎科生物有限公司测序,获得pAbAi-BpGLK1-E1重组质粒。
1.2.2 靶点设计及gRNA的合成
利用CRISPR-direct网页(http://crispr.dbcls.jp/)筛选特异靶点。在BpGLK1第1外显子BpGLK1-E1(图1)选定1个靶点序列,根据靶点序列合成BpGLK1-gRNA引物(表1),采用PCR技术合成gRNA的双链DNA模板。反应体系为:rTaq1 μL、10×PCR Buffer(Roche)5 μL (5 U/μL)、BpGLK1-gRNA引物2 μL(0.4 μmol/L)、BS6 2 μL(500 mmol/L)、T25-long 5 μL(10 μmol/L)、BS7 10 μL(10 μmol/L)、dNTP Mix 2 μL(2.5 mmol/L),加ddH2O补足体积50 μL。反应程序为:95 ℃预变性1 min,95 ℃变性10 s,59℃退火10 s,72℃延伸10 s,40个循环,72 ℃ 30 s,16 ℃保温,反应结束即可获得gRNA双链DNA。随后用T7快速RNA合成试剂盒合成gRNA,反应体系中分别加入:NTP Buffer Mix 7.5 μL、BpGLK1-gRNA双链DNA 8 μL、T7 RNA聚合酶1.5 μL,加水至总体积20 μL,充分混匀后37 ℃反应4 h,即可获得BpGLK1-gRNA。

表1 gRNA靶序列合成相关引物

下划线序列为靶位点序列 The underlined sequence is the target site sequence。图1 BpGLK1第1外显子序列Fig. 1 BpGLK1 exon 1 sequence
1.2.3 CRISPR/Cas9核糖核蛋白(RNP)体外活性检测
以pAbAi-BpGLK1-E1质粒(长度为5 840 bp)为模板(图2),用BstEⅡ酶切后获得线性化质粒,随后进行体外活性检测。活性检测反应体系:2 μL Cas9蛋白、2 μL gRNA、12 μL线性化的pAbAi-BpGLK1-E1质粒、0.8 μL 1 mol/L(pH 7.5)磷酸缓冲液、1 μL(100 mmol/L) MgCl2、0.02 μL(1 mol/L)dDTT充分混匀,37 ℃下孵育1 h,1%(质量分数,下同)琼脂糖凝胶电泳检测,根据线性化pAbAi-BpGLK1-E1质粒能否被剪切成两条谱带(其中1条谱带长度为BpGLK1-gRNA靶点到BstEII限制酶切位点约1 155 bp左右,另1条线性化质粒为剩余部分约为4 685 bp),判断Cas9蛋白活性及gRNA的精准性。
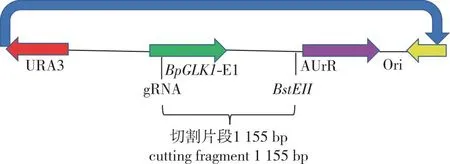
图2 pAbAi-BpGLK1-E1载体简图Fig. 2 pAbAi-BpGLK1-E1 vector diagram
1.2.4 基因枪微弹的制备及轰击白桦愈伤组织
将白桦种子用流水冲泡72 h后,在超净工作台中进行消毒,随后纵向切割种子,剥去种皮,将其接种到愈伤诱导培养基(WPM+2.0 mg/L 6-BA+0.2 mg/L NAA)中置于无光照条件下培养,20 d左右即可获得幼嫩的愈伤组织,用于基因枪法轰击受体。根据相关文献[15]取30 mg金粉加入Eppendorf管,加入1 mL 70%(体积分数)酒精,充分涡旋,静置15 min,1 500 r/min离心5 min。弃上清,加入1 mL无菌水,涡旋充分,1 500 r/min离心5 min,进行3次重复。加入1 mL的50%甘油,此时终浓度达到了60 mg/mL,4 ℃保存。CRISPR/Cas9 Ribonucleoprotein(RNP)的包裹,涡旋金粉母液5 min以打碎成团的微粒。需要注意的是,微粒很容易下沉,每一枪吸取的时候都要重新振荡重悬微粒。取50 μL 金粉悬液于1.5 mL Eppendorf管。依次加入CRISPR/Cas9 RNP复合物,在室温(25 ℃)下将5 μL Cas9蛋白和5 μL gRNA(gRNA体外验证已获得)混合,总体积为10 μL,50 μL 2.5 mol/L CaCl2、20 μL 0.1 mol/L亚精胺(现用现配,边涡旋边加入,每加完一样试剂,振荡2~3 s)。上述混好的样品,涡旋1 min,冰上静置1 min,重复3次。静置10 min,10 000 r/min离心10 s,去上清。200 μL无菌水冲洗沉淀2次,10 000 r/min离心10 s,去上清。后用40 μL无菌水重悬微粒。制备包被Cas9蛋白+gRNA的基因枪微弹,采用PDS-1000/He基因枪(Bio-Rad, 美国),爆破压力为7 584.5 kPa,轰击距离为12 cm,进行白桦愈伤组织的轰击。轰击结束后将愈伤组织接种于分化培养基(WPM +1.0 mg/L 6-BA+0.02 mg/L NAA+0.5 mg/L GA3)中,置于无光照条件下3~5 d后,随后在1 500~2 000 lx条件下培养25 d左右,待愈伤组织产生不定芽后转接于继代培养基(WPM +1.0 mg/L 6-BA+0.02 mg/L NAA),3 000~4 000 lx条件下培养25 d左右,每25 d更换继代培养基。剪取高为1 cm左右的不定芽转接到生根培养基(WPM+0.4 mg/L IBA)中进行生根培养。上述培养基中均添加200 g/L蔗糖及5.5 g/L琼脂,培养室温度为(25±2)℃。
1.2.5 基因编辑株系的分子检测
分别提取基因编辑株系组培生根苗的DNA, 以基因编辑株系叶片总DNA为模板,用特异引物BpGLK1-gRNA-F.CTCTCCAGTCTTCCTTCCGAT,BpGLK1-gRNA-R.CTGAAGGTGGCTGGCTATGT进行PCR扩增,用1%的琼脂糖凝胶对PCR产物进行检测,并送往擎科生物有限公司测序,通过Bioedit软件将测序结果与基因组序列进行比对。
2 结果与分析
2.1 pAbAi- BpGLK1-E1重组载体的获得
以白桦基因组DNA为模板,用BpGLK1-E1两端特异引物进行PCR扩增,1%琼脂糖凝胶电泳检测显示,在1 000 bp处可见扩增谱带(图3a),与预期的BpGLK1第1外显子946 bp长度吻合。进而采用双酶切技术分别对上述扩增片段及pAbAi质粒进行双酶切,经T4酶连接后并转化大肠杆菌,挑取单克隆进行PCR检测,凝胶电泳结果显示,在1 000 bp处可见扩增谱带,与预期的946 bp长度吻合(图3b),将pAbAi-BpGLK1-E1重组质粒送往擎科生物公司测序,经Bioedit比对后显示BpGLK1-E1序列正确,可用于后续实验。
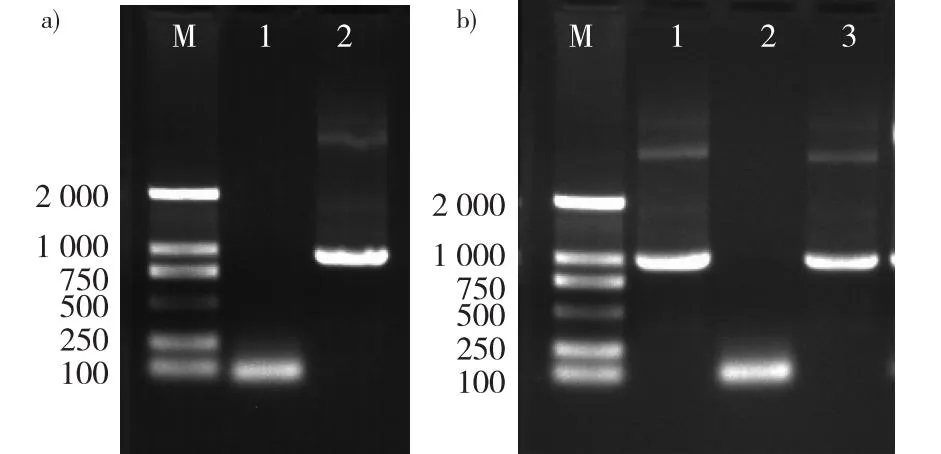
a. BpGLK1-E1靶序列扩增电泳图electrophoretogram of BpGLK1-E1 target sequence amplification;M.Marker DL2000;1.水water;2. BpGLK1-E1序列 BpGLK1-E1 sequence;b.重组质粒检测PCR扩增电泳图谱 electropherogram of recombinant plasmid PCR detection;M.Marker DL2000;1. BpGLK1质粒 BpGLK1plasmid;2.水water;3. pAbAi-BpGLK1-E1质粒 pAbAi-BpGLK1-E1 plasmid。图3 pAbAi-BpGLK1-E1重组载体获得Fig. 3 Acquisition of pAbAi-BpGLK1-E1 recombinant vector
2.2 Cas9蛋白的核酸酶活性
以pAbAi-BpGLK1-E1质粒载体为模板,采用BstEⅡ限制酶酶切得到线性化重组质粒。进而以线性pAbAi-BpGLK1-E1重组质粒为模板(含有BpGLK1靶序列),在含Cas9蛋白及gRNA的缓冲液中反应后(同时设置3组阴性对照:不加Cas9蛋白、不加gRNA、空载体),琼脂糖凝胶电泳检测结果见图4。
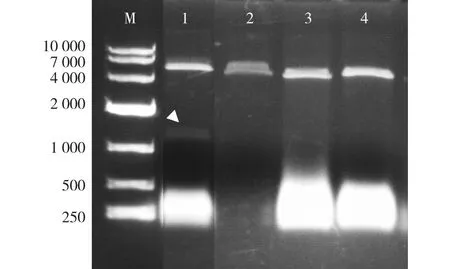
M.DL10000;1. gRNA-Cas9蛋白复合物 gRNA-Cas9 protein complex; 2. 无Cas9蛋白对照 no Cas9 protein;3. 空载(阴性对照)empty (negative control);4. 无gRNA对照no gRNA control。图4 CRISPPR/Cas9靶位点活性检测Fig. 4 Activity detection of CRISPPR/Cas9 target site
由图4可知,1 155 bp左右可检测到电泳谱带,而3个对照组在1 155 bp左右则未见电泳谱带,表明Cas9蛋白及gRNA复合体具有裂解活性。
2.3 白桦glkc基因编辑株系的获得
以白桦成熟胚为材料,在无光照条件下进行愈伤组织诱导,25 d左右即可用于基因枪轰击的受体。在基因枪参数设置为7 584.5 kPa爆破压力,轰击距离12 cm的条件下,用包被Cas9蛋白与gRNA(RNP)金粒轰击白桦幼嫩愈伤组织,在分化培养过程中愈伤组织渐渐膨大(图5a),随后转接到继代培养基中培养25 d左右时,可见长高的不定芽。在继代培养的不定芽中明显可见叶色呈现黄绿色的编辑株系(命名为glkc)(图5b),待30~40 d时与野生型白桦呈现明显差异(图5c、5d)。

a.愈伤组织callus;b.继代培养的 glkc株系differentiated cultured glkc line;c.野生型白桦wild type;d. glkc株系glkc line。图5 白桦glkc基因编辑株系的获得Fig. 5 Acquisition of glkc gene editing line
2.4 glkc株系的分子检测
以glkc株系的10株组培生根苗叶片总DNA为模板,用BpGLK1-gRNA-F、BpGLK1-gRNA-F特异引物对BpGLK1-E1片段进行PCR扩增。琼脂糖凝胶电泳检测显示:glkc株系在900 bp左右有扩增谱带(图6a),进而将扩增谱带回收后对其中5株进行测序。测序结果通过Bioedit软件比对,发现测序结果一致,glkc编辑株系在BpGLK1基因的315~332 bp处有18个碱基缺失的纯合突变(图6b),结果说明,获得的glkc株系为BpGLK1缺失突变株。
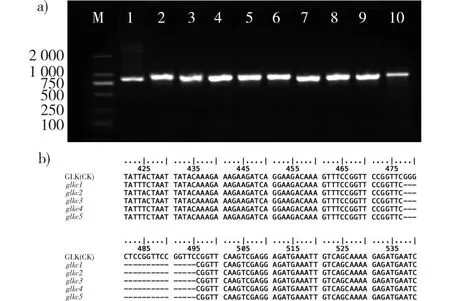
a.BpGLK1-E1序列扩增电泳图谱electrophoretogram of BpGLK1-E1 target sequence amplification;M.Marker DL2000; 1—10. glkc株系组培生根苗 rooted seedling of glkc line by tissue culture; b.glkc基因编辑株系测序比对结果sequencing comparison result of glkc gene editing line。图6 白桦glkc株系的分子检测Fig. 6 Molecular detection of glkc line
3 讨 论
近年来兴起的CRISPR/Cas9 系统由于能够精准进行基因编辑,已在基因工程领域广泛应用。以往人们主要采用质粒转染或直接转染体外转录的Cas9 mRNA及gRNA等方法将Cas9蛋白和gRNA导入细胞,其中,质粒转染可能导致外源基因(T-DNA或抗性基因)的插入[10];而直接转染Cas9 mRNA 和gRNA的方式虽然避免了外源基因插入的发生,但存在着RNA易降解等问题[16]。RNP技术是近年兴起的直接将Cas9蛋白和gRNA复合物导入细胞的新技术,即Cas9蛋白与gRNA在试管中孵育,通过静电互作组装形成Cas9/gRNA复合物(RNP)[17],导入细胞后即可发挥其基因编辑功能。RNP技术中体外表达的Cas9蛋白的活性和gRNA的精准性非常重要,因此,通常在进行基因编辑前需要对其进行活性检测。本研究检测显示,体外表达的Cas9蛋白具有活性,gRNA也具有定点编辑功能。
植物中主要采用微粒轰击或聚乙二醇(PEG)将Cas9/gRNA 复合物导入细胞,例如,Woo等[10]通过转染CRISPR/Cas9 RNPs进入原生质体,获得了基因组修饰的莴苣(Lactucasativa)。在拟南芥、烟草、矮牵牛(Petuniahybrid)、葡萄藤和苹果的原生质体中也获得了基因编辑突变体,但没有再生突变植株[18-19]。白桦作为树木,相对模式植物及农作物而言,研究比较滞后,从原生质体再生植物仍然是一个挑战。另外,对于一些大粒种子的小麦(Triticumaestivum)、玉米等作物通常以未成熟胚为受体,利用微粒轰击将CRISPR/Cas的核糖蛋白RNP导入植物组织细胞中[9, 20]。按前人经验采用种子胚是一条有效途径,而白桦种粒较小(长宽仅为1.5和1 mm),去除种皮操作过程较费时,故此,本研究通过将少量的种子胚在暗培养条件下进行愈伤诱导,培养获得大量的松散愈伤组织,以此愈伤组织为受体利用基因枪将CRISPR/Cas核糖蛋白(RNP)微粒导入白桦细胞中,经过分化培养,即可获得白桦glkc编辑株系。本研究利用CRISPR/Cas9核糖核蛋白的无外源基因整合的基因编辑系统,以白桦合子胚诱导的愈伤组织为受体,采用微粒轰击技术进行BpGLK1定向诱变,获得了glkc编辑突变株,为创制林木无T-DNA插入突变体提供了新线索。该项研究优势在于这种无T-DNA插入的无选择标记的转化方法,不需要经过有性杂交和自交将转入的CRISPR/Cas9系统和编辑的靶位点编辑基因从个体中分开,可以在T0代中生成不含外源DNA的经过编辑的基因编辑株系。
研究证明,GLK基因家族成员在调控植物的叶绿体发育及叶绿素合成等方面至关重要,例如,拟南芥AtGLK1和AtGLK2双突变体叶片表现为叶绿素含量降低,呈现黄色[21],在番茄(Solanumlycopersicum)中SIGLK1抑制表达株系其叶色呈现黄绿色,其叶绿素含量较对照株系降低[22]。对白桦、银中杨(Populusalba×P.berolinensis)的研究也显示,BpGLK1基因的缺失及干扰表达可通过影响叶绿体发育进而导致叶片叶绿素含量降低,叶片呈现鲜艳黄绿色[12-13, 23]。glkc突变株则不同,该株系通过CRISPR/Cas9编辑后,在BpGLK1基因的315~332 bp处有18个碱基的缺失,是由于碱基的缺失而导致BpGLK1基因编码蛋白功能发生改变,该基因缺失突变产生新的未知蛋白,但从获得的glkc编辑株系生长来看,这种突变是不利突变,glkc突变株的组培无根苗在生根培养基中生根能力较弱,不定根数量较少,在生根过程中苗木叶片渐渐变为白色,最后叶片干枯后衰老死亡。BpGLK1基因序列分析显示,该基因共有6个外显子,本实验是在第1外显子处设计的靶位点,后续还需在其他5个外显子处设计靶位点,找到不影响白桦突变株系生长的编辑靶位点。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
亚热带植物科学(2022年1期)2022-05-17
中国生殖健康(2020年4期)2021-01-18
文苑(2019年20期)2019-11-20
文苑·经典美文(2019年10期)2019-10-16
四川农业科技(2019年5期)2019-07-01
中国生殖健康(2018年4期)2018-11-06
华人时刊(2016年16期)2016-04-05
浙江柑橘(2016年1期)2016-03-11
湖北农业科学(2014年11期)2014-09-10
