
基于转录组测序的关联分析定位裂叶桦叶形调控基因
2024-01-23 05:35顾宸瑞袁启航穆怀志刘桂丰
南京林业大学学报(自然科学版) 2024年1期
顾宸瑞,袁启航,姜 静,穆怀志,刘桂丰*
(1. 林木遗传育种全国重点实验室(东北林业大学),黑龙江 哈尔滨 150040;2. 北华大学林学院, 吉林 吉林 132013)
作为欧洲白桦(Betulapendula)的变种,裂叶桦(B.pendula‘Dalecarlica’)叶片开裂成掌状,顶部叶尖细长,一级齿减少,叶脉在叶背面明显隆起,内部机械输导组织非常发达,且横切面积显著增大,叶背绒毛发达,在园艺观赏、育种工作及植物发育生物学研究中具有重要的应用价值,但其叶片形态变异机制及调控基因始终未被揭示[1-2]。
双子叶植物的叶片性状呈现非常丰富的多样性,一般从叶形指数、叶缘缺刻程度和叶缘锯齿3个方面对叶片的形状进行衡量,而这3个指标分别受叶侧脉延伸速率和叶肉蹼化程度两个因素的控制,叶侧脉延伸速率大则叶片较宽,叶肉的蹼化程度在整个叶缘较低则出现锯齿,蹼化过程在某几个叶脉间的位置受抑制则出现掌状裂[3]。
植物的叶形可能是数量性状或质量性状,其受环境影响严重,且某些植物存在异形叶性,决定叶形的成因复杂,这给定位控制叶形性状的主效应基因带来一定难度[4]。目前对双子叶植物叶片形状的多样性及其遗传规律的研究主要集中于棉花(Gossypiumhirsutum)、羽衣甘蓝(Brassicaoleraceavar.acephala)、白菜(Brassicarapevar.glabra)等叶面积能够直接或间接影响产量的作物上。其中,影响棉花叶片形状的基因已被定位于其第15号染色体[5],GhLMI1-like和GhKNOX1基因对叶形的控制已被验证[6];羽衣甘蓝的叶缘缺刻性状被认为是数量性状,但起决定性的主效应基因共有2对[7-9];白菜不结球裂叶突变体的裂叶性状被发现是受一对显性基因控制的质量性状,并已发现与之关联的遗传标记[10]。
关联分析以连锁不平衡为基础,利用基因组上的分子标记,定位特定性状对应的基因座。随着高通量测序技术的发展,全基因组关联分析(GWAS)已成为研究群体数量性状变异的常用手段[11-14]。GWAS策略利用规模较大的野生群体和全基因组高密度SNP标记定位目标基因座,可节省群体构建成本和时间。但这种关联分析需要对目标性状进行精确的测量和量化,而叶形不规则程度(复叶,裂片、叶缘锯齿等特征)缺乏合适的量化标准,对这些数量性状的关联分析几乎没有报道;另一方面,GWAS使用DNA重测序数据,不能同时检测基因的变异和表达量差异,而植物叶片的形态建成往往受基因表达量影响[15]。转录组测序可一次性获取外显子区的SNP标记和基因表达量信息,但受限于无法检测基因间区SNP标记的缺点,基于转录组测序的关联分析未被广泛采用。
近年来,在裂叶桦与欧洲白桦的杂交实验中,裂叶桦叶形始终被作为质量性状进行统计[2],但孟德尔分离规律不能解释子代群体中出现的中间过渡型个体和超亲遗传个体。本研究提出了一种量化叶形不规则程度的指标,计量了24株不同叶形白桦的叶不规则程度,并与其RNA-Seq数据进行关联分析,旨在定位控制白桦叶形的主效应基因座,预测候选基因,为后续的验证和育种工作奠定基础。
1 材料与方法
1.1 供试材料
试验材料来自裂叶桦(中裂叶)×欧洲白桦的一个半同胞家系子代,定植于东北林业大学白桦强化种子园。该家系有卵形叶、深裂叶、中裂叶和浅裂叶4种叶形的后代(图1),每种选取6株,共24株,用于后续研究。

图1 实验材料不同类型叶片示例Fig. 1 Leaf shape examples for experimental material
1.2 试验方法
1.2.1 叶不规则度的测量和比较
深裂叶类型每株摘取3片成熟功能叶,其他叶形每株随机摘取5片发育成熟的功能叶,共108片叶。使用Epson Expression 10000XL扫描仪(日本)获取叶片图像,所有图片以300 dpi进行扫描。
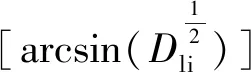
1.2.2 影响裂叶桦叶形基因的定位
24个植株顶端分生组织RNA-Seq数据来自本团队的前期工作[16]。使用RNA-STAR软件[17]将测序数据比对到欧洲白桦参考基因组(CoGe:35080)[18],使用sambamba软件[19]去除重复读段,使用GATK4分析SNP/InDel,并将24个植株的SNP/InDel信息汇总。使用beagle软件[20-21]进行数据填补。使用plink软件[22]进行过滤,标准为:MAF≥5%,GENO≤20%,MIND≤20%,PHWE≥0.001(MAF表示第2等位基因频率,GENO表示位点基因型缺失比例,MIND表示样本基因型缺失比例,PHWE表示哈迪温伯格平衡显著性)。使用Tassel 5.0软件[23]进行基于一般线性模型(GLM)的关联分析。以P=0.01/n计算Bonferroni 阈值(n为参与关联分析的SNP/InDel个数)筛选目标SNP。
1.2.3 候选基因及注释
根据参考基因组注释文件查找目标SNP覆盖的基因。使用htseq-count软件[24]依据RNA-Seq数据计算全基因组基因表达量,使用DESeq2软件[25]计算基因表达差异显著性(将来自6株正常叶个体的数据视为野生型的6次生物学重复,将来自18株裂叶个体的数据视为裂叶桦的18次生物学重复),并筛选出位于关联区的差异基因。使用COG、Gene Ontology、KEGG、KOG、Pfam、Swissprot、TrEMBL、NCBI-nr等在线数据库对候选基因进行注释。
2 结果与分析
2.1 实验材料表型数据
根据裂叶桦×欧洲白桦(B.pendula)半同胞家系24株子代共108片成熟叶的图像计算叶不规则程度,并进行方差分析(表1)和多重比较(表2)。

表1 叶形不规则度方差分析结果

表2 个体间叶形不规则程度多重比较结果
由表1可见,叶不规则程度在不同个体之间差异极显著(P<0.01)。由表2可见,叶形不规则程度变幅较大,不规则指数最高可达0.352 2,最低仅0.032 8,平均0.182 4。样本变异系数为60.51%,满足进行关联分析的条件。多重比较结果表明,该量化指标在不同叶形个体间差异显著,其数值能够代表不同个体的叶形。
2.2 关联分析及候选基因的预测与注释
RNA-Seq数据系来自Illumina HiSeq 2500平台的双端100 bP第2代高通量测序结果,共331 Gb,平均每个样品13.8 Gb。过滤后,Q30比例均在90.62%以上,与参考基因组比对率均在85.36%以上。
由RNA-Seq数据共获得SNP/InDel 3 303 530个,经过补齐和筛选,保留了高质量多态SNP/InDel 1 367 359个。采用GLM模型进行关联分析,发现超过Bonferroni 阈值(-lgP≥ 8.135 883)的候选SNP/InDel共66个(图2 a、2 b)。由于实验材料来自同一半同胞家系,将零星出现于3、6、11号染色体上的5个候选SNP视为异常值剔除。
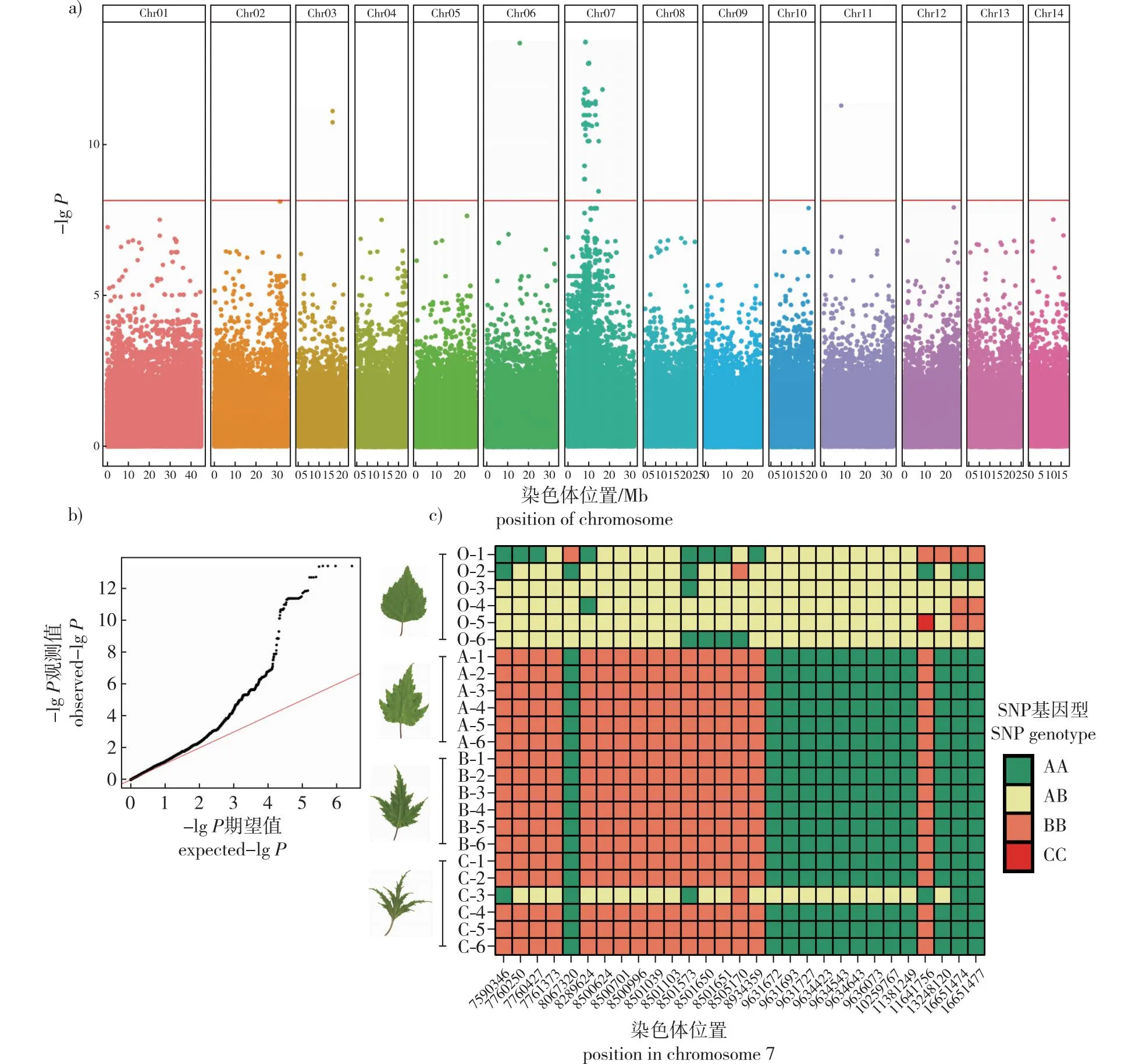
a. 曼哈顿图Manhattan plots; b.QQ图 Quantile-quantile plots;c.7号染色体上目标SNP在个体中的基因型 genotypes of aim SNPs on chromosome7。图2 叶片不规则程度关联分析结果Fig. 2 Association analysis result of leaf irregularity degree
保留的61个目标SNP/InDel分布于一个9.19 MbP的关联区中(BpChr07:7466344—16651477)。该区域共包含400个基因,其中17个基因的编码区携带上述目标SNP/InDel。位于基因编码区的SNP基因型均不能与表型完全关联,但这些SNP普遍在正常叶个体与裂叶个体之间基因型存在差异,除深裂叶个体C-3外,目标SNP在裂叶桦中基因型一致(图2 c)。
24个样品的全基因组基因表达量及差异显著性由DESeq2分析,在关联区中,表达量在裂叶个体与正常叶个体之间存在极显著差异(P<0.01)的基因共25个,其中6个在裂叶桦中上调表达,19个在裂叶桦中下调表达(图3 a)。
被目标SNP影响编码的基因和关联区差异表达基因共计40个,其中2个同时具有SNP和表达量差异(图3 b)。使用公共数据库对候选基因进行了功能注释,在表3中,对注释结果进行了概括性的描述。

表3 候选基因的变异类型及注释信息
由表3可见,候选基因中包括转录因子3个、作为细胞组成成分的蛋白3个、参与蛋白质翻译加工的蛋白6个、参与信号传导的蛋白激酶9个、参与生化反应的酶5个、参与物质运输的蛋白3个、对RNA进行转录后修饰的蛋白1个,DNA内/外切酶1个,以及调节气孔的蛋白1个。另外,8个基因无法在现有的在线数据库中查询到,其类型与功能完全未知。
3 讨 论
3.1 Dli(4πS/C2)可用作量化植物叶形不规则程度的指标
植物叶片边缘的形态通常以“锯齿”“裂叶”“深裂”“复叶”等非量化指标进行描述[15],而缺少将叶片形态的不规则程度进行量化的指标,因此,植物的叶形变异通常被作为质量性状处理,或被解释为异形叶性。本研究所使用的半同胞家系,个体出现从卵形叶到鸡爪状深裂叶的分布,为了进行关联分析,需要对叶缘开裂程度进行量化。本研究以Dli作为衡量叶片不规则程度的指标,该指标为平面图形面积与周长相等的圆的面积之比。
值得注意的是,在计算叶片图像周长时,会受到尺缩效应的影响,即图片精细度不足以展示叶缘锯齿时,会使测得的周长偏小;而叶缘绒毛、破损等细节被过度放大时,会使测得的周长偏大。因此对每一批次的比较,必须先通过预实验选定适用于该物种的像素密度,而后在相同的像素密度下扫描样本图像。
3.2 基于转录组数据进行关联分析的优势与不足
随着高通量测序技术的发展,以GWAS为代表的利用高密度SNP为标记的关联分析方法被广泛应用于数量性状基因座的定位。通常,GWAS所需的样本来自大规模的野生群体,数量在100以上,样本的数量性状表型值呈连续的正态分布[26]。
本研究中,对24株来自同一半同胞家系的材料进行转录组测序,并通过一般线性模型(GLM)进行了关联分析。由于实验材料来自一个半同胞家系,且样本数量较少,这使得实际P值过低,高于阈值的SNP过多,观测-lgP值总体偏高,不服从正态分布,因而不能简单地套用GWAS的分析策略。本研究表明,在半同胞家系中,相邻的SNP标记体现出连锁效应。笔者效仿BSR分析的策略[27],引入关联区的概念,一方面将关联区外零星出现的高于阈值的SNP视为异常值剔除,另一方面对关联区中所有发生了可能与表型有关的变异(包括关联性SNP/InDel与差异表达)的基因进行注释。结果表明,调控裂叶桦叶形的基因坐落于一个9.18MbP的关联区,其中40个基因出现了可与表型相关联的编码变异或表达量差异。
为了避免第一类错误,笔者汇总了全部40个在不同叶形个体之间存在差别的基因,统一进行了基因功能注释。但本研究未检测到与表型完全关联的SNP,这可能是由于裂叶桦的叶形是多个基因突变的综合调控结果,或由某基因的启动子变异导致。能够一次性获得序列变异和表达量差异是RNA-Seq相较于DNA-Seq的优势,但RNA-Seq不能检测基因启动子的序列,因此对差异表达的基因,应在后续研究中关注其启动子是否存在变异。
3.3 关联分析结果为基因精细定位奠定了基础
候选基因中共包含3个转录因子,其中2个隶属于MYB家族(Bpev01.c1499.g0007、Bpev01.c1097.g0004),1个隶属于WOX家族(Bpev01.c1525.g0003)。在拟南芥(Arabidopsisthaliana)中,AS1作为MYB结构域蛋白,可通过与KNOX等转录因子拮抗或互作调控叶片近/远轴的形态建成从而影响叶形[28];在烟草(Nicotianasylvestris)中,WUS作为WOX家族成员,参与分生组织的维持,是叶原基形成和叶脉建成所必需的[29]。候选基因中的一个PIN家族蛋白(Bpev01.c0147.g0002)在白桦(B.platyphylla)[30]中的同源基因BpPIN1已被克隆,BpPIN1的表达量与白桦叶片发育过程紧密相关,并在白桦与裂叶桦之间存在显著表达模式差异[31]。在拟南芥中,PIN1作为介导生长素极性运输的外泌蛋白,直接制造叶原基内部特定位置的生长素浓度峰点,继而诱导这些位置发育成叶脉[32]。裂叶桦裂叶性状的成因可能与上述4个候选基因有关,但不能排除其他候选基因参与调控叶形的可能,尤其是8个未能在现有数据库中查询到的候选基因。这8个未知基因可能是白桦所特有的,暂无法对其功能进行预测。
本研究所用材料来自一个半同胞家系,由于此类近缘群体中相邻SNP的连锁较强,导致定位结构所得关联区较大。使用近缘群体进行关联分析的同类研究案例表明,此类关联分析是可行的,其结合精细定位工作成功确定了目的基因[33]。精细定位可缩小关联区范围,大量地排除因近缘群体SNP连锁及随机阳性信号带来的第二类错误,排除与叶形无关的候选基因。在后续研究中,可使用分子生物学手段,借助基因组上已发掘的SSR标记[18, 30]和本研究中检测到的SNP标记,在本研究基础上开展精细定位工作,将关联区进一步缩小。
猜你喜欢
新世纪智能(数学备考)(2021年9期)2021-11-24
文苑(2019年20期)2019-11-20
文苑·经典美文(2019年10期)2019-10-16
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
读者(2017年5期)2017-02-15
华人时刊(2016年16期)2016-04-05
语文教学与研究(读写天地)(2014年2期)2014-05-26
语文教学与研究(2014年6期)2014-03-12
湖南农业科学(2014年10期)2014-02-27
