
Fe/Co/Ni-N共掺杂石墨烯氧还原反应活性的DFT研究
2024-01-18 13:37马俊杰刘建峰袁斌霞潘卫国施正荣
原子与分子物理学报 2024年3期
马俊杰, 宁 锴, 王 婷, 刘建峰, 袁斌霞, 潘卫国, 施正荣,2
(1. 上海电力大学 能源与机械工程学院, 上海 200090; 2.上海非碳基能源转换与利用研究院,上海200240)
1 引 言
由于全球的能源短缺和环境污染,寻找替代化石燃料的解决方法正成为一项重要任务.燃料电池作为一种环境友好、高效的势能转换装置,受到了广泛的关注,在燃料电池阴极上发生的氧还原反应(ORR)决定了燃料电池的电化学性能和能量转换效率[1].然而氧还原反应的进行在运动上是比较缓慢的,转换效率是不高的,因此,氧还原活性和高效的阴极催化剂对提高燃料电池的能量转换效率和电化学性能具有重要的意义.目前,最好的阴极催化剂仍然是铂(Pt)贵金属,它被用作高活性电催化剂,但这种材料的高价格、稀缺资源、和低稳定性和限制了其广泛的工业应用[2].近几年来,大量的电化学专家和学者为了取代或减少Pt催化剂作为燃料电池阴极的使用,集中精力开发用于燃料电池阴极的新材料,因此,非贵金属燃料电池阴极催化剂受到了广泛的关注. 由于碳具有较高的导电性、可负载性、相对较高的化学稳定性和电化学稳定性,被认为是质子交换膜燃料电池阴极催化剂的最佳负载材料之一,石墨烯正是一种新型的碳基材料[3].
随着社会发展和科技进步,新型材料的研究被人们广泛关注,2004年,英国曼彻斯特大学的Andre Geim和Constantine Novoselov从石墨中分离出石墨烯[4],新型碳材料石墨烯的出现由于其优异的结构特性和性能带来了新的研究热潮.石墨烯具有独特的电学特性,作为催化剂载体有着良好的氧还原活性,在催化领域有着广阔的应用前景[5].但由于石墨烯的规则性结构和半导体属性限制了其被调控的空间,因此石墨烯可以通过化学或者物理方法进行修饰或改性,这可以改善石墨烯的性质,拓宽石墨烯的应用领域. 杂原子和非贵金属对石墨烯化学掺杂可以改变石墨烯的内部结构,打开石墨烯的能带带隙,从而有效的提升石墨烯的性能.合适的掺杂原子还能改变石墨烯的电化学性能和催化性能[6].由于非贵金属成本低,活性高,所以非贵金属原子掺杂的石墨烯被广泛应用于提高氧还原活性,非贵金属(铁、钴、和镍等)和氮共掺杂碳(M-Nx-C)催化剂对氧还原反应具有良好的催化活性[7].目前有研究表明,非贵金属钴和杂原子氮共掺杂碳催化剂(Co-Nx-C)具有良好的ORR活性[8];另外锰和氮共掺杂碳催化剂(Mn-N4-C)也表现出较高的ORR活性[9];铁和氮共掺杂碳催化剂(Fe-N4-C)的理论研究表明,与Pt相比,它对ORR具有较高的催化活性[10].然而人们对非贵金属(铁、钴、和镍等)和不同氮共掺杂石墨烯(M-Nx-G)催化剂的ORR活性的研究还不够全面,对其对氧气的吸附和脱附的探索还不够深入,因此本文依据密度泛函理论(DFT)对铁、钴、镍等非贵金属和不同氮共掺杂石墨烯催化剂的ORR活性进行了研究,并着重分析了催化剂对氧气的吸附、脱附和催化剂的导电性.
2 模型的建立和计算方法
2.1 模型的建立
石墨烯是以六个杂化碳原子构成的单原子层二维晶体结构,因此使用Materials Studio软件建立周期平板模型,以模板库中的“graphite”为重复单元,建立4×4×1的超胞,共有41个碳原子,考虑到镜像间的相互作用,真空层取15 Å.在单金属原子掺杂石墨烯时存在两种构型M-C3和M-C4,为了比较Fe,Co,Ni掺杂石墨烯时存在的两种构型的稳定性和ORR活性,构建了单金属掺杂石墨烯的两种构型,如图1(a-b)所示.单金属原子和氮共掺杂石墨烯也有两种构型,但是M-C3构型不稳定,因此在单金属和氮共掺杂时均采用和M-C4构型,为了比较Fe,Co,Ni和N共掺杂石墨烯(M-Nx-G)的ORR活性,构建了单金属和氮共掺杂石墨烯的4种构型,如图1(c-f),分别为M-N1-G、M-N2-G、M-N3-G和M-N4-G.然而M-N2-G构型又存在3种不同构型,分别为M-N21-G、M-N22-G和M-N23-G,如图1(g-i),这样能更直接的发现不同构型对氮掺杂的影响.在本文中,单金属原子M指的是Fe,Co,Ni之一.
2.2 计算方法
本文基于第一性原理的密度泛函理论,应用Materials Studio软件进行全部的模拟计算.使用Materials Studio软件的CASTEP模块对建立的氮掺杂石墨烯模型进行结构优化,选择几何优化为任务目标,使用广义梯度近似理论(GGA-PBE)交换关联泛函的方法进行模拟计算,由于整个Fe,Co,Ni和N共掺杂石墨烯模型是致密的体系结构,所以使用这种方法得到的计算结果更为精确[11].设定相应的截断能为400 eV,收敛质量为中等,总能量的收敛精度为2.0×10-5eV/Å,每个原子最大的力的收敛精度为0.05 eV/Å,最大位移为0.002 Å,最大迭代步数为100,考虑自旋极化效应,使用正常自旋为初始自旋.布里渊区积分采用Monkhorst-Pack型网格,K点取值为2×2×1.为了比较Fe,Co,Ni和N共掺杂石墨烯模型体系的吸附能和脱附能,选择能带结构、态密度和布局分析进行运算.
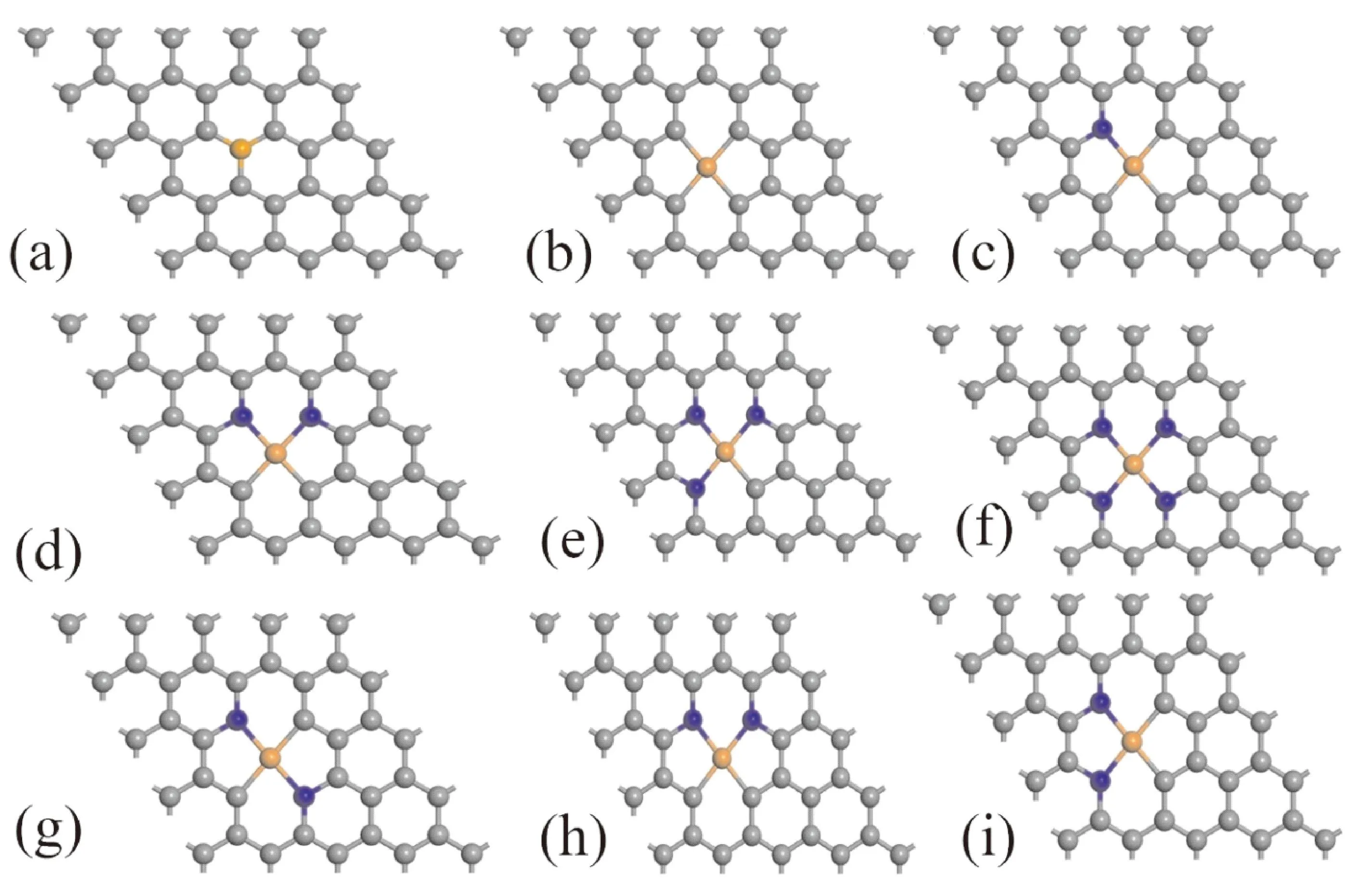
图 1 单金属掺杂石墨烯构型(a-b)、单金属和氮共掺杂石墨烯构型(c-f)和 M-N2-G的3种构型(g-i)Fig. 1 Monometallic doped graphene configurations (a-b),monometallic and nitrogen co-doped graphene configurations (c-f)and three configurations of M-N2- G (g-i)
Materials Studio软件是研究原子级多孔材料结构和性质的有效工具,提供了全面的模拟环境,集量子力学、分子力学和介观模拟为一体,可以解决催化剂、化学反应等材料和化学领域的一些重要问题[12].其中的CASTEP模块有独特的以DFT为基础的量子力学程序,一般多应用于均相催化研究、多相催化、分子反应研究等方面,以使用原子中心网格的数值函数作为原子基[13],其基函数由不同原子的DFT方程得到,这样可以准确的描述体系.由于CASTEP模块的优化方法相较于其他模块更加的准确而且高效,多以该模块成为了进行分子DFT研究(尤其是周期性分子)的精度最高的模块之一.
3 结果与讨论
3.1 吸附能
理论和实验研究表明,吸附能是催化剂氧还原反应活性的有效描述,吸附能与氧还原反应活性的关系对催化剂的性能具有重要的意义[14].由于吸附能是一个作为展现电化学耦合的参数,而氧还原反应活性归结于界面电子耦合,因此吸附能也是用来定义表面反应物的氧还原反应活性的一个很好的筛选因子[15].固体催化剂的表面活性是由每个活性位点的整体活性(本征)和暴露的活性位点数(外在)决定的,而电催化的氧还原反应过程包含了催化剂表面反应物的吸附过程,在反应物和催化剂之间打断某些结合键来形成新的化学键,从而得到活性中间体[16].
Fe,Co,Ni和N共掺杂石墨烯吸附氧分子时,会存在物理吸附和化学吸附,但是化学吸附起主导作用,因此本文只研究化学吸附.通过GGA-PW91方法对体系进行弛豫,得到吸附氧分子的稳定构型,再分别分析各个体系的吸附氧气的相关性质.本文定义体系的吸附能计算公式如下:
Eadsorption=Etotal-Emolecule+Esurface
在M-Nx-G吸附氧气模型中,Emolecule为氧分子的能量,Esurface为M-Nx-G的能量,Etotal为体系总能量,也就是氧分子能量与M-Nx-G能量之和.吸附能反映了界面两边物质相互作用的强弱,如果吸附能是负值,则绝对值越大,相互作用力也越大.
Fe,Co,Ni掺杂石墨烯时,由于M-C3和M-C4两种构型的体系总能量相差不大,因此为了论证的严谨性,对这两种构型对氧气的吸附能都进行了计算.在单金属掺杂石墨烯中,因为金属的强还原性,氧气分子会直接与金属原子发生化学吸附并形成离子键.氧分子可以以OOH、O和OH的结构形式吸附在石墨烯表面,因为OOH的吸附形式最为常见,据此选择OOH的吸附形式,如图2(a-b),M-C3和M-C4吸附氧气之后的构型分别为M-G-A和M-G-B.经过模拟计算,Fe,Co,Ni掺杂石墨烯分别吸附氧气的吸附能如表1所示.从表中可以看出,无论是M-G-A还是M-G-B构型,都是Fe掺杂石墨烯吸附氧气的吸附能最大,达到0.19 eV和0.37 eV,这说明氧分子更容易与Fe相结合,更有利于电子的转移;而且在M-G-B构型中,Fe掺杂石墨烯吸附氧气的吸附能要远远大于Co和Ni.
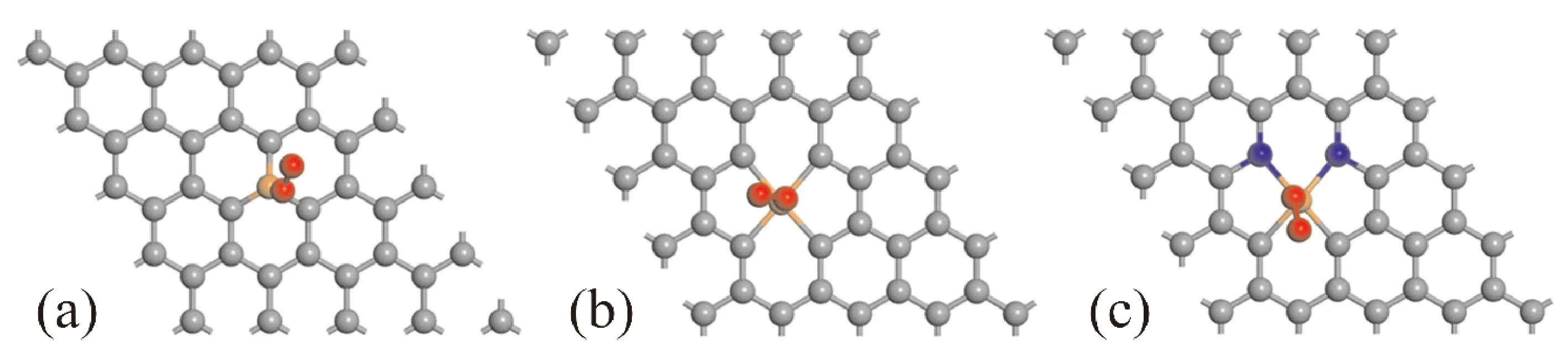
图2 氧分子在M-G-A(a)、M-G-B(b)和M-N22-G(c)上的稳定吸附构型Fig. 2 Stable adsorption configurations of oxygen molecules on M-G-A (a),M-G-B (b)and M-N22-G (c)

表1 Fe,Co,Ni掺杂石墨烯吸附氧气的吸附能
Fe,Co,Ni和N共掺杂石墨烯时,氮的掺杂一共有6种,分别为M-N1-G、M-N21-G、M-N22-G、M-N23-G、M-N3-G和M-N4-G,其吸附氧气时同样是以OOH的结构形式吸附在石墨烯表面,氧分子与金属原子吸附成键,其中M-N22-G的构型如图2(c)所示.模拟计算后,Fe,Co,Ni和N共掺杂石墨烯分别吸附氧气的吸附能如表2所示.由表可知,M-N4-G催化剂的氧气吸附能最大,这应该是氮含量对其ORR活性产生了影响;在M- N4-G构型中,Fe-N4-G吸附氧气的吸附能又大于Co-N4-G和Ni-N4-G,达到3.74 eV,说明Fe与N共掺杂石墨烯的氧气吸附更容易;而M-N21-G、M-N22-G和M-N23-G吸附氧气的吸附能相差并不明显,因此M-N2-G的三种构型对其ORR活性的影响几乎相同.

表2 Fe,Co,Ni和N共掺杂石墨烯吸附氧气的吸附能
3.2 脱附能
化学吸附并不是氧还原反应活性高低的唯一判断标准,化学脱附同样是氧气还原反应过程中的一个重要环节[17].氧气从掺杂石墨烯基底脱附的脱附能决定着氧分子是否容易从掺杂石墨烯中脱附,完成电子的转移,因此研究氧分子在掺杂石墨烯表面的脱附,对于催化反应的机理和判断氧还原反应活性具有十分重要的意义.本文通过GGA-PW91方法对Fe,Co,Ni和N共掺杂石墨烯体系进行弛豫,得到氧分子脱附中各个过程的稳定构型,再分别分析各个体系的氧分子脱附的相关性质.
氧分子从Fe,Co,Ni掺杂石墨烯脱附时,由于氧分子是以OOH的结构形式吸附在石墨烯表面,氧分子的脱附会经历OOH、OH和H2O的过程,如图3(a-c)所示.经过模拟计算之后,再分别计算氧分子变为OOH、OH和H2O三个状态的能量变化,最后算出脱附能,如表1所示.从表1中可以看出,Fe-G-A、Co-G-A和Ni-G-A脱附氧气的脱附能相差不大,说明Fe,Co,Ni掺杂石墨烯以M-C3的形式掺杂时其脱附能几乎相同;Fe-G-B脱附氧气的脱附能最小,仅为3.06 eV,说明Fe掺杂石墨烯以M-C4的形式掺杂时,氧分子更容易脱附,更有利于电子转移.氧分子从Fe,Co,Ni和N共掺杂石墨烯脱附时,氧分子的脱附同样会经历OOH、OH和H2O的过程,如图3(d-e)所示.模拟计算后,再分别计算氧分子变为OOH、OH和H2O三个状态的能量变化,最后得到脱附能,如表2所示.从中可以看出,在M-N4-G构型中,Fe-N4-G脱附氧气的吸脱附能比Co-N4-G和Ni-N4-G更小,为1.77 eV,说明Fe和N共掺杂石墨烯催化剂氧气脱附过程更容易发生;但总体来看,Fe-Nx-G脱附氧气的脱附能相差并不明显,说明氮含量对Fe和N共掺杂石墨烯的ORR活性影响不大,Co-Nx-G和Ni-Nx-G也同样是如此.

图3 氧分子从M-G-B(a-c)和M-N1-G(d-f)中脱附的过程Fig. 3 Desorptions of oxygen molecules from M-G-B (a-c)and M-N1-G (d-f)

表3 Fe,Co,Ni掺杂石墨烯脱附氧气的脱附能
表4 Fe,Co,Ni和N共掺杂石墨烯脱附氧气的脱附能
3.2 导电性
导电性是描述催化剂氧还原活性的另一个有效参数[18].通过GGA-PBE方法分别计算出Fe-Nx-G的能带结构,得到Fe和N共掺杂石墨烯的带隙,如表5所示.从表中可以看出,随着氮含量增加,Fe和N共掺杂石墨烯的带隙也越来越大,其中Fe-N4-G的带隙最大,为0.341 eV,因此Fe-N4-G的导电性也最好,说明氮含量的增加有利于打开石墨烯结构的带隙,使得其导电性变好.而Fe-N21-G、Fe-N22-G和Fe-N23-G的带隙相差并不明显,因此Fe-N2-G的三种构型对其导电性的影响几乎相同.

表5 Fe和N共掺杂石墨烯的带隙
4 结 论
基于密度泛函理论模拟研究了Fe,Co,Ni和N共掺杂石墨烯的吸附能、脱附能和导电性,从而比较单金属原子和氮不同的共掺杂方式对石墨烯ORR活性的影响.基于模拟结果分析,从吸附能来看,Fe掺杂石墨烯和Fe和N共掺杂石墨烯吸附氧气的吸附能最大,更容易吸附氧气,且Fe-N4-G吸附氧气的吸附能在Fe-NX-G中最大;从脱附能来看,Fe以M-C4的形式掺杂石墨烯时,氧分子脱附更容易进行,且Fe和N共掺杂石墨烯更容易使氧气脱附;从导电性的角度分析,氮含量的增加有利于增强石墨烯的导电性.综上所述,同时考虑吸附能、脱附能和导电性,单金属原子掺杂石墨烯时,Fe掺杂石墨烯的ORR活性优于Co和Ni;单金属原子和氮共掺杂石墨烯时,M-N4-G的ORR活性优于M-N1-G、M-N2-G和M-N3-G,且Fe和N共掺杂石墨烯的ORR活性优于Co和Ni.
猜你喜欢
军事文摘(2023年22期)2023-12-19
中学生数理化·中考版(2022年9期)2022-10-25
中学生数理化·中考版(2021年9期)2021-11-20
纺织科学研究(2021年7期)2021-08-14
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
北京航空航天大学学报(2017年10期)2017-04-20
儿童故事画报·自然探秘(2016年6期)2016-09-14
中国塑料(2016年3期)2016-06-15
浙江大学学报(工学版)(2016年9期)2016-06-05
电源技术(2015年5期)2015-08-22
