
穿流式电催化膜MnO2@Ti 的制备及催化氧化正丙醇性能
2024-01-15 11:00杨明霞樊森清陈皎皎肖泽仪
化学反应工程与工艺 2023年5期
杨明霞,樊森清,陈皎皎,陈 渝,李 创,肖泽仪
四川大学化学工程学院,四川 成都 610065
丙酸是重要的精细化工中间体。目前丙酸的主要化学合成路线有丙醛氧化和乙烯羰基化,但存在反应条件苛刻,工艺流程复杂,催化剂有毒等不足[1-3]。正丙醇来源广、安全性高、价格便宜,进一步脱氢后可生成丙酸,被认为是生产丙酸有前途的前驱体。电化学反应通过在温和条件下以可控方式产生高活性自由基从而实现反应物的转化[4],是可以替代使用氧化剂或还原剂等对环境有害的化学试剂的化学过程,通过正丙醇电催化氧化制备丙酸符合绿色化学发展理念。
传统电极通常将粉末状电催化剂负载在电极表面,这种结构会导致催化剂团聚掩埋活性位点。将催化剂原位固定在多孔导电膜的膜孔中构成电催化膜,较大的膜孔表面积有利于提高纳米催化颗粒的负载量,且分散良好的膜孔可以防止催化剂聚集超过膜孔尺寸,从而增强反应物和催化剂接触[5-6]。传统电催化反应通常是在间歇模式下进行,其电化学氧化动力学通常受到质量传递的限制,因为相对于电子传递而言化学扩散更缓慢。如果反应流体可以通过强制对流的方式与膜孔中的催化剂接触,电催化反应将被限制在微-纳尺度的孔道内,极大地缩短传质距离,提高电催化反应速率[7-10]。Liu 等[11]研究表明,与间歇反应相比,穿流式电化学反应器的传质速率增加了六倍。此外,由于均匀的膜孔分布,正丙醇在膜中的停留时间分布均匀,正丙醛等副反应将被削弱,预计会得到更高的丙酸选择性。
在前期的研究中[12-15],已通过流动合成法成功地在膜孔中原位固载了催化剂纳米颗粒,催化剂前体溶液在外力作用下均匀缓慢渗透过膜,使得纳米颗粒在膜厚度方向均匀分布,构筑了功能催化膜。根据多孔介质深层过滤的原理,由于惯性碰撞和布朗运动,电催化纳米颗粒可以稳定固载在曲折的膜孔中。本研究将通过流动合成法在微米级钛膜孔道中原位固载MnO2纳米催化剂,构筑MnO2@Ti 电催化膜,并在新设计的一种穿流式电催化氧化装置中研究MnO2@Ti 电催化膜催化正丙醇选择性氧化生成丙酸的性能,进一步探讨其氧化机理。
1 实验部分
1.1 实验原料
多孔管式钛膜购自南京雄凯过滤设备有限公司,内径和外径分别为20 mm 和25 mm,平均孔径为1 μm,孔隙率为22%,高度为60 mm。管式膜的一端用不锈钢板密封,另一端用不锈钢接头密封作为连接蠕动泵软管的出口。将钛膜置于10 g/L 的草酸溶液中超声预处理30 min 以除去表面的杂质和对膜表面进行刻蚀,然后用大量去离子水清洗至溶液为中性,在60 ℃的烘箱中烘干备用。
正丙醇,丙酸,正丙醛和异丁醇为色谱纯。硫酸锰一水合物、高锰酸钾、硫酸钠和草酸为分析纯,所有试剂无需进一步纯化即可使用。
1.2 流动合成法制备电催化膜
首先,通过蠕动泵加压使0.8 mol/L 硫酸锰溶液缓慢匀速渗透过钛膜25 min,在60 ℃下干燥3 h,使得硫酸锰溶液在膜孔中逐渐饱和并结晶。然后,以同样方式将0.2 mol/L 高锰酸钾溶液均匀渗透过膜45 min,在这一过程中,高锰酸钾溶液与膜孔中的硫酸锰发生氧化还原反应合成MnO2,将膜在60 ℃下干燥3 h。重复以上两个步骤,增加负载次数,使MnO2纳米颗粒在膜孔中逐渐积累,从而调整MnO2负载量。最后,超声清洗除去膜表面稳定性差的MnO2纳米颗粒以及溶解膜内未反应的溶液,在60 ℃下烘干。
1.3 正丙醇电催化氧化实验
穿流模式下的电催化氧化反应装置如图1 所示。MnO2@Ti 电催化膜被用作阳极,环绕在阳极周围的不锈钢网作为阴极。稳压直流电源提供恒定电流,并用导线连接两个电极形成电路。蠕动泵提供恒定的负压,使正丙醇溶液以一定的速率流经MnO2@Ti电催化膜进行循环氧化。在实验过程中,正丙醇水溶液为反应物,0.11 mol/L 的硫酸钠溶液为电解质。在穿流模式下测试了MnO2@Ti 电催化膜的电催化性能,每个样品测量三次并计算平均值以减少误差。为了研究MnO2@Ti 电催化膜在穿流模式下的增强机制,将MnO2@Ti 电催化膜进行了无流动的间歇模式下的正丙醇电催化氧化实验,间歇模式和穿流模式的实验条件相同。

图1 MnO2@Ti 电催化膜在穿流模式下正丙醇电催化氧化实验装置Fig.1 Experimental device for the electrocatalytic oxidation of n-propanol by MnO2@Ti electrocatalytic membrane via flow-through mode
1.4 分析测试
X 射线衍射(XRD)图像由X 射线衍射仪(Empyrean)测得,以分析MnO2的晶体结构,X 射线源为Cu-Kα,管电压为50 kV。原始Ti 膜和MnO2@Ti 电催化膜的表面和横截面的微观结构采用场发射扫描电子显微镜(SEM,JEOL,JSM-700F)进行观察。通过电子顺磁共振(EPR,Bruker EMX plus X-ban)对产生的羟基自由基(·OH)进行测量,中心磁场为3 510 G,扫描宽度为100 G,微波频率为9.86 GHz。通过CHI604E 电化学工作站进行了循环伏安法(CV)、电化学阻抗谱(EIS)和电流密度-时间曲线(I-t)测定。采用三电极体系,MnO2@Ti 电催化膜或原始Ti 膜被用作工作电极,铂片被用作对电极,Ag/AgCl 电极被用作参比电极。CV 曲线扫描速率为5 mV/s。EIS 在开路电压下测得,频率扫描为0.01~100 000 Hz,幅度为50 mV,CV 曲线和EIS 测量是在0.11 mol/L 的Na2SO4电解质溶液中进行。I-t曲线在电流密度为2.55 mA/cm2的条件下测定,溶液由160 mmol/L 正丙醇和0.11 mol/L的Na2SO4电解质溶液组成。反应物和产物用气相色谱仪(GC-9790Ⅱ,浙江福立分析仪器股份有限公司)进行分析,FFAP 色谱柱(30 m×0.32 mm×1.0 μm),氢火焰离子化检测器(FID),载气为氮气,流速为24 mL/min,检测器和汽化室温度设置为250 ℃,柱箱温度设为程序升温,初始温度设为60 ℃,停留时间为1 min,以10 ℃/min 的速率升至150 ℃,保温5 min。
正丙醇氧化生成丙酸反应为液相反应,正丙醇转化率(xn-propanol)、丙酸的选择性(SCOOH)和丙酸电流效率(CECOOH)按式(1)~式(3)计算:
式中:C0和Ct分别为MnO2@Ti 电催化膜反应前后正丙醇浓度,mol/L;Ct-COOH为反应时间为t时溶液中的丙酸浓度,mol/L;nCOOH为生成的丙酸的量,mol/m2;zCOOH为正丙醇产生的丙酸的电子转移数,为4;F为法拉第常数,为96 500 C/mol;I为电流密度,A/m2;t为反应时间,h。
2 结果与讨论
2.1 电催化膜的结构和形貌
MnO2@Ti 电催化膜的XRD 图谱见图2。从图2可以看出,流动合成法制备的MnO2在2θ为37.2°,42.4°,56.0°和66.7°处的衍射峰分别对应于(100),(101),(102)和(110)晶面,与JADE 标准图谱中六方相ε-MnO2(JCPDS 30-0820)的特征峰相对应,表明通过流动合成法在膜孔中成功合成了ε-MnO2[16]。
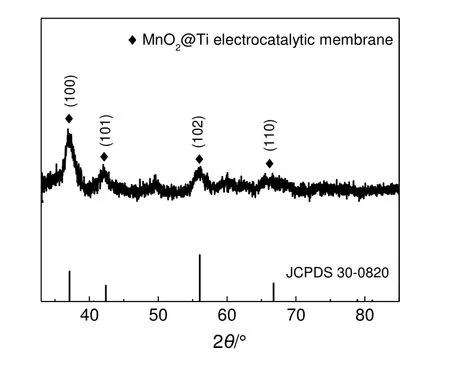
图2 MnO2@Ti 电催化膜的XRD 图谱Fig.2 XRD pattern of MnO2@Ti electrocatalytic membrane
原始钛膜、MnO2@Ti 电催化膜的表面和横截面的SEM 照片,以及其中的纳米颗粒的尺寸分布见图3。由SEM 照片可以看出,原始钛膜表面及孔道内壁光滑,经过流动合成后,球形的MnO2纳米颗粒在膜孔道中均匀分布,且平均粒径为83.1 nm。在膜表面也存在部分MnO2纳米颗粒,且平均粒径为246.4 nm,约为孔道中的催化剂粒径的3 倍。与膜表面合成的MnO2相比,膜孔内合成的尺寸更加均一,这是因为流动合成可强化两种反应物之间的传质,减少反应物消耗造成的浓度差。

图3 原始钛膜(a, b)和MnO2@Ti 电催化膜(c)的膜表面SEM;表面MnO2 粒径分布(d);原始钛膜(e, f)和MnO2@Ti 电催化膜(g)的横截面SEM;横截面MnO2 粒径分布(h)Fig.3 SEM images of the membrane surface of the Ti membrane (a, b) and MnO2@Ti electrocatalytic membrane (c);MnO2 particle size distribution on Surface of electrocatalytic membrane (d);SEM images of the cross-section of the Ti membrane (e, f) and MnO2@Ti electrocatalytic membrane (g);MnO2 Particle size distribution along the cross-section of electrocatalytic membrane (h)
2.2 电催化膜的电化学性能
分别测试了原始钛膜和MnO2@Ti 电催化膜的循环伏安曲线,图4(a)显示了两种膜的CV 曲线。可以看出,与原始钛膜相比,MnO2@Ti 电催化膜的电流密度显著提高。MnO2@Ti 电催化膜在1 VvsAg/AgCl(相对于参比电极的电位)的电位下显示出高达3.8 mA/cm2的峰值电流密度,而原始钛膜在Na2SO4溶液中峰值电流密度仅为0.22 mA/cm2。电流密度的增加表明MnO2@Ti 电催化膜电极在该体系中具有快速的电子迁移过程。这是因为MnO2@Ti 电催化膜的多孔结构极大缩短了离子传输途径,且大的表面积有利于电荷传输。
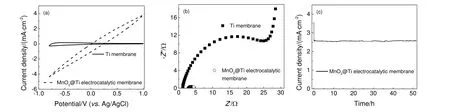
图4 钛膜和MnO2@Ti 电催化膜的循环伏安曲线(a),电化学阻抗谱拟合曲线(b)和I-t 曲线(c)Fig.4 Cyclic voltammograms (a), electrochemical impedance spectra fitting curves (b) and I-t curve (c) of the Ti membrane and the MnO2@Ti electrocatalytic membrane
采用EIS 测试了电极各部分电阻的变化,图4(b)展示了钛膜与MnO2@Ti 电催化膜的电化学阻抗谱的拟合曲线(阻抗的实部Z’和虚部Z”)。可以看出,奈奎斯特图在与界面电荷转移电阻(Rct)相关的高频区域(图左侧区域)显示为半圆形,与质量扩散行为有关的低频区(图右侧区域)为线性。通过Zview 软件拟合得到电催化膜电极的Rct值为1.4 Ω,远低于原始钛膜的Rct值(27.5 Ω),表明MnO2@Ti 电催化膜的法拉第过程或电荷转移动力学更快[17-18]。这归因于MnO2@Ti 电催化膜的多孔结构和较高的表面积可以缩短电荷的扩散距离以及强化传质,有利于电子在MnO2上快速转移。EIS 结果证明在流动合成MnO2纳米粒子后,电极的电化学性能显著提高,这与CV 测量结果一致。
通过I-t曲线对MnO2@Ti 电催化膜的稳定性进行了研究,结果如图4(c)所示。结果表明,在电流密度为2.55 mA/cm2的条件下,在50 h 内电催化膜作为阳极表现出良好的电催化稳定性,其衰减可以忽略不计。其良好的稳定性是因为流动合成制备的电极通过前驱体溶液连续的冲洗可以去除电极孔隙中不稳定的催化剂。
2.3 MnO2@Ti 电催化膜电催化氧化正丙醇性能
2.3.1 反应模式的影响
为了探讨MnO2@Ti 电催化膜作为阳极对正丙醇选择性氧化的强化行为,考察了相同条件下MnO2@Ti 电催化膜在穿流模式以及间歇反应模式下正丙醇电催化氧化反应性能,结果见图5。由图5可见,间歇反应模式时反应19 h,MnO2@Ti 电催化膜催化正丙醇的转化率仅为60%,丙酸的选择性为70%,而穿流模式下正丙醇的转化率高达95%,丙酸的选择性提高到89%。此外,MnO2@Ti 电催化膜在间歇模式下的电流效率为44%,在穿流模式下提高到90%。因为在穿流模式下正丙醇氧化过程中的传质是在MnO2@Ti 电催化膜微米尺度的膜孔空间内进行,反应物与催化剂表面的传质距离仅限于膜孔的大小,这比反应溶液平行与电极表面流动的传统电极的传质距离缩小了几个数量级,强化了传质过程。在穿流模式下丙酸选择性的提高是因为电催化膜中均匀的膜孔尺寸的分布为正丙醇的氧化提供了均匀的水力停留时间,水力停留时间是从膜上游经过膜孔到膜下游的时间,这在传统的平行于电极表面流动的结构中是不可能实现的。而穿流模式下更高的正丙醇转化率和丙酸选择性意味着更高的电流效率。结果表明,电催化过程限域效应与流动协同强化了传质,显著提高了正丙醇氧化性能。
2.3.2 反应条件的影响
不同条件下MnO2@Ti 电催化膜在穿流模式下正丙醇的电催化氧化反应结果见图6。如图6(a)所示,在反应时间为14 h 的条件下,随着初始正丙醇浓度的增加,MnO2@Ti 电催化膜对正丙醇的处理能力先是增加,然后趋于平缓,而正丙醇转化率逐渐降低,丙酸的选择性有部分下降。这是因为一定的电流密度产生的羟基自由基是恒定的,催化剂上有足够的活性位点和羟基自由基会限制正丙醇的氧化过程,不再将正丙醛中间物进一步氧化成丙酸。此外,当正丙醇初始浓度较高时,更多正丙醇和不能及时氧化的中间体会积累在电极表面,使传质受到阻碍,降低了单位时间内单分子与活性位点接触的几率[19-21]。

图6 不同反应条件下MnO2@Ti 电催化膜氧化正丙醇的性能Fig.6 Performance of n-propanol oxidation by MnO2@Ti electrocatalytic membrane under different initial concentrations of n-propanol
电流密度是电化学有机合成的重要参数,也是电催化速率的关键影响因素[22]。如图6(b)所示,在反应时间为14 h 的条件下,当电流密度从1.27 mA/cm2增加到3.80 mA/cm2,MnO2@Ti 电催化膜对正丙醇的处理能力、正丙醇转化率和丙酸的选择性均增加,但进一步增加电流密度则对正丙醇的处理能力、正丙醇转化率和丙酸选择性的影响不大。这是因为随着电流密度增加,电极的氧化电位增大可以促进正丙醇的氧化反应,但是较高的电流密度会导致阳极发生严重的电解水析氧取代产生羟基自由基,且电解水产生的大量气泡会阻碍正丙醇与催化膜中的活性位点接触,从而阻碍正丙醇的氧化。
MnO2的负载量对正丙醇处理能力、正丙醇转化率以及丙酸的选择性的影响如图6(c)所示。可以看出,在相同反应时间下,随着MnO2负载量的增加,正丙醇的转化率以及对正丙醇的处理能力均增加,而丙酸的选择性基本不变。不同MnO2负载次数所得催化剂的负载量和膜通量如表1 所示。由表1 可知,随着负载次数增加,MnO2的负载量是成倍增加的,但膜通量降低。因此,较高的MnO2负载量可以提供更多的电催化活性位点,从而获得更好的催化性能。

表1 MnO2@Ti 电催化膜中MnO2负载量和膜通量Table 1 MnO2 loading and membrane flux in MnO2@Tielectrocatalytic membrane
2.4 MnO2@Ti 电催化膜氧化正丙醇制备丙酸的机理
在电催化氧化过程中可能会产生许多种类的自由基[23],可以通过EPR 来验证电催化氧化过程中产生自由基的种类。实验中使用5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)对·OH 进行捕捉,当DMPO浓度为0.1 mol/L,电解质为0.11 mol/L 的Na2SO4,电流密度为2.55 mA/cm2时,MnO2@Ti 电催化膜通电120 s 以及没有外加电流时产生自由基的EPR 图谱见图7。由图7 可以看出,通电120 s 时DMPO捕捉后生成的自由基加合物产生了明显的1:2:2:1 对称结构的四重峰,表明MnO2@Ti 电催化膜在电催化氧化过程中产生了·OH,其作为强氧化基团在电催化氧化反应过程中有着重要的作用[24]。正丙醇电催化氧化产物已经通过气相色谱确定,中间物和产物分别为正丙醛和丙酸,因此提出了MnO2@Ti 电催化膜氧化正丙醇生成丙酸可能的机理,如图8 所示。

图7 MnO2@Ti 电催化膜产生自由基的EPR 谱图Fig.7 EPR spectrum for free radicals generated by MnO2@Ti electrocatalytic membrane
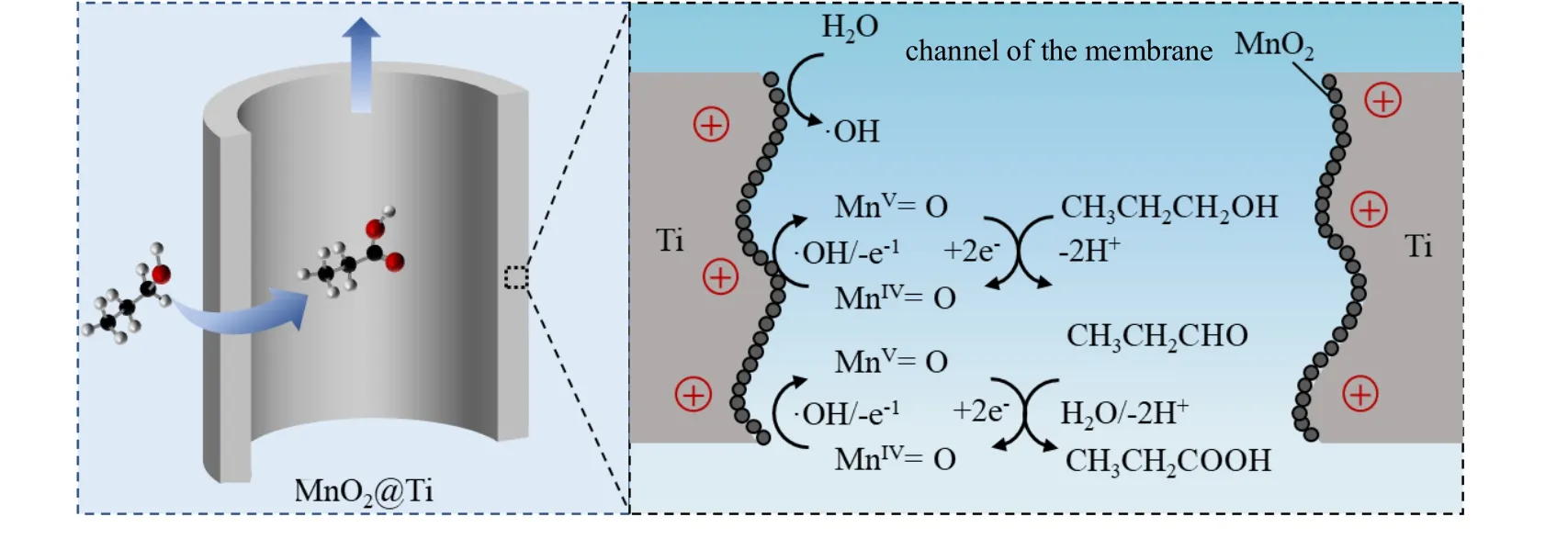
图8 MnO2@Ti 电催化膜催化氧化正丙醇可能的反应机理Fig.8 Possible reaction mechanism of n-propanol oxidation over the MnO2@Ti electrocatalytic membrane
首先,含有金属氧化物电极上发生水氧化产生吸附的羟基自由基,见式(5)。羟基自由基进一步将MnIV=O 转化为MnⅤ=O 进而演变成化学吸附氧,见式(6)。MnO2催化剂具有多种高价态的锰氧化物,通常是强化学氧化剂。通过MnO2的电子转移可以不断生成MnⅤ=O,吸附在表面的正丙醇会被MnⅤ=O 氧化生成中间产物正丙醛,而MnⅤ=O 将转化为MnIV=O,见式(7),同时H+会被传递到阴极,在阴极与电子结合生成H2,见式(8)。正丙醛则通过氧化还原反应进一步被氧化成丙酸,见式(9)。所以MnO2@Ti 电催化膜催化氧化正丙醇制备丙酸的反应路径可以如式(10)描述。
MnO2@Ti 电催化膜在穿流模式下对正丙醇进行电催化氧化反应过程中,产生的短寿命·OH 将会被限制在微米级的膜孔中[25-26]。由于·OH 的寿命极短,穿流模式下快速的传质过程可以增强吸附在催化剂表面的短寿命·OH 与正丙醇的接触[27],使得更多的·OH 可以被利用,降低·OH 的淬灭速度。因此,MnO2@Ti 电催化膜在穿流模式下具有更好的正丙醇氧化性能,且实现了更高的电流效率。
3 结 论
通过流动合成法制备了用于正丙醇氧化的MnO2@Ti 电催化膜。电化学表征表明MnO2@Ti 电催化膜的电化学性能可以显著提高,且保持长时间的稳定性。MnO2@Ti 电催化膜在穿流模式下展现出优异的催化正丙醇氧化制备丙酸的性能,正丙醇的转化率可以达到95%,丙酸的选择性可达到89%,阳极的电流效率可以达到90%。考察了不同操作条件对正丙醇电催化氧化反应的影响,结果表明通过适当提高反应物的浓度、电流密度和催化剂的MnO2负载量有利于正丙醇氧化生成丙酸反应。对正丙醇电催化氧化生成丙酸的机理分析表明,穿流模式下强化接触和传质能使电催化膜氧化过程中更多的·OH 被利用,从而提高正丙醇的氧化性能。
猜你喜欢
酿酒科技(2023年9期)2023-11-25
石油化工(2020年8期)2020-09-15
食品工业科技(2018年18期)2018-10-16
中国有色金属学报(2018年2期)2018-03-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
浙江大学学报(工学版)(2016年2期)2016-06-05
中国资源综合利用(2016年7期)2016-02-03
化工学报(2015年1期)2015-04-01
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
石油化工应用(2014年6期)2014-03-11
