
Oxygen vacancies enriched Ni-Co/SiO2@CeO2 redox catalyst for cycling methane partial oxidation and CO2 splitting
2024-01-13 04:53ChangYangJupingZhangJiakaiWangDongfangLiKongzhaiLiXingZhu
Chang Yang, Juping Zhang, Jiakai Wang, Dongfang Li, Kongzhai Li, Xing Zhu
State Key Laboratory of Complex Nonferrous Metal Resources Clean Utilization, Faculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming 650093, China
Keywords: Chemical looping Methane Dry reforming Catalyst Partial Oxidation
ABSTRACT Redox catalysts play a vital role in the interconversion of two significant greenhouse gases,CO2 and CH4,via chemical looping methane dry reforming technology.Herein,a series of transition metals-alloyed and core–shell structured Ni-M/SiO2@CeO2 (M = Fe, Co, Cu, Mn, Zr) redox catalyst were fabricated and evaluated in a gas–solid fixed-bed reactor for cycling CH4 partial oxidation (POx) and CO2 splitting. The catalysts are composed of spherical SiO2 core and CeO2 shell, and the highly dispersed Ni alloy nanoparticles are the interlayer between core and shell.The oxygen vacancy concentration of Ni-M/SiO2@CeO2 followed the order of Co>Cu>Fe>Mn >Zr,and Ni alloying with transition metals significantly enhanced oxygen storage capacity (OSC). Ni-Co/SiO2@CeO2 catalyst with abundant oxygen vacancies and a high OSC showed the lowest temperatures of CH4 activation (610 °C) and CO2 decomposition (590 °C), thus demonstrating excellent redox reactivity.The catalyst exhibited superior activity and structural stability in the continuous CH4/CO2 redox cycles at 615°C,achieving 87%CH4 conversion and 83%CO selectivity.The proposed catalyst shows great potential for the utilization of CH4 and CO2 in a redox mode,providing a new sight for design redox catalyst in chemical looping or related fields.
1. Introduction
CO2and CH4are two important greenhouse gases that can be utilized as carbon resources, making their resource utilization of great significance [1–3]. Dry reforming of methane (DRM), which leverages the reducing capacity of CH4and the oxidizing capacity of CO2to achieve CO2—CH4interconversion, is a promising approach to reduce carbon emission [4,5]. However, due to the stable molecular structures of CO2and CH4, dry reforming usually requires a high operating temperature of 700–900 °C [6,7]. Meanwhile,the co-feeding mode of CO2—CH4results in the limited conversion rate of the two raw materials due to thermodynamic equilibrium [8,9]. Furthermore, the theoretical value of H2/CO molar ratio of syngas produced by dry reforming is 1, which is not optimal for subsequent conversion and utilization. Thus, it is clear that developing a new dry reforming technology to improve the mutual conversion efficiency of CO2—CH4is necessary.
In contrast to the co-feeding mode, chemical looping DRM(CL-DRM) technology takes advantage of the characteristics of chemical looping process [10,11]. CH4and CO2are fed separately instead of co-feeding, thus avoiding the limitations imposed by the equilibrium conversion rate in the co-feeding mode and enabling product separation. Moreover, synthesis gas and pure CO gas products with H2/CO molar ratios suitable for liquid fuel synthesis can be obtained [12]. CL-DRM usually divides the interconversion of CO2—CH4into two gas–solid reaction steps: reduction and oxidation. First, CH4is selectively oxidized by redox catalyst (also known as oxygen carrier) in the reduction step to produce synthesis gas, and then CO2is decomposed by reduced redox catalyst to produce CO in the oxidation step. Redox steps are alternately carried out to realize the continuous conversion of CH4and CO2[13]. This reaction mode also facilitate the activation of CO2and CH4, thus reducing the reaction temperature [14]. The key of the CL-DRM process is the redox catalyst,which is required to be capable of activating carbon species and transferring lattice oxygen.
Several factors should be taken into consideration when designing redox catalysts, such as reactive performance, cyclic stability,cost and environmental impact [15,16]. Although noble metals have demonstrated good low-temperature activity in dry reforming[17],they are not preferred due to their scarcity and high cost.Most studies focus on non-noble metals that display high catalytic performance and are low-cost [18]. At present, transition metals such as Ni-based, Fe-based, Cu-based, Co-based and composite metal oxides(perovskite oxygen carriers[19],CeO2oxygen carriers[20,21],hexaaluminate oxygen carriers[13],etc.)have been widely reported as redox catalysts using in chemical looping conversions of methane and CO2. Among these, transition metal oxides exhibit high methane reaction activity and valence changing ability. Ni[22,23]and Co[24]oxides have excellent methane activation ability, while Cu [25] and Mn [26] exhibit high CH4complete oxidation, and Fe oxides [27] have high oxygen storage capacity (OSC).In addition,numerous studies show that Ni/CeO2exhibits superior CH4/CO2activation ability in chemical looping reforming.
Highly dispersed Ni-based catalysts can activate CH4and facilitate the migration of lattice oxygen [1]. Meanwhile, CeO2is an excellent carrier of nickel-based catalyst, as well as a promising container for oxygen storage. CeO2also could increase the Ni dispersion and exhibit a good catalytic performance for CH4—CO2conversion. These effects are originated from the fluorite structure of CeO2based catalyst and the plentiful oxygen vacancies generated from Ce4+/Ce3+transformation[12].During the redox process,oxygen vacancies can enhance the oxygen transfer and increase the oxygen releasing/acquiring ability of the catalyst, which provides an efficient way for surface reaction to transport oxygen through the bulk lattice and creates effective surface sites for reactant adsorption and subsequent activation [28]. The O2–migration velocity and OSC of ceria could be strengthened by the addition of transition metal ions into lattice. The abundant oxygen vacancies and high OSC can enhance the oxygen storage/release ability of the catalyst. Marinet al. [29] adopted CeO2-Fe2O3as the carrier of Ni and oxygen storage material for chemical looping conversion of CO2and found that the addition of Ni can greatly increase the CO yield. Similarly, Löfberget al. [30] investigated the reactivity of Ni/CeO2in CL-DRM of methane and found that Ni played a crucial role in the activation of reactants. Ni-based and Ce-based catalysts show excellent performance in CL-DRM. However, these catalysts are prone to deactivation due to sintering of Ni at high temperature. Most researches focus the conversion of methane into syngas at temperature ranged from 550 to 700 °C to achieve a favorable cycling stability of catalysts. However, the conversion of methane is low in this temperature range. Zhuet al. [31] doped Ni on CeZrO2to activate methane, and the activation temperature of CH4was reduced to enable the selective oxidation of methane into syngas at 650 °C. The process produced a syngas with H2/CO molar ratio close to the ideal ratio of 2.0 and the CH4conversion was over 90%. Chenet al. [32] applied a Ni-CeZrAl catalyst in DMR and obtained a methane conversion of 88% at 650 °C. They also observed that the presence of abundant basic sites and oxygen vacancies helped to form disordered carbon to protect the active Ni active sites.Tsaiet al.[33]developed a bifunctional material composed of CaO-Ni-CeO2by combining calcium cycle with DRM at 680 °C for the generation of syngas with high yields. Thus, it is expected to achieve more efficient conversion of CH4and CO2at lower temperature by structural design of the catalyst.
Plenty of studies have proven that the carbon resistance of catalysts can be improved by increasing the metal-carrier interaction,doping metal oxides, or fabricating specific microstructure [34].Wanget al. [20,35] found that the addition a small amount of Ni to CeO2can improve the performance of CL-DRM. They attributed this excellent performance to Ni nanoparticles coated by CeO2through strong metal-carrier interaction (SMSI), which created more surface oxygen vacancies for CH4activation.Liet al.[36]synthesized a solid solution based catalyst composed of NiO-MgO and CeO2-NiO by multi-step co-precipitation method. The addition of NiO created strong interactions between the Ni species and CeO2-MgO to activate the carriers and oxygen vacancies,which significantly improved the conversion rate of CH4and the yield of CO.The synergistic effect of bimetals enhances the dispersibility of the catalyst and provides additional active sites required on the surface of the catalyst.The development and utilization of bimetallic catalysts facilitate the coupling of the synergistic redox of different metals,the increasing of active site density on the catalyst surface.The alloyed to mixed valence transition metals to bimetallic materials is a convenient and effective way to obtain catalysts with high activity, high stability and low economy. Liet al. [37] prepared a core–shell structured Fe2O3@LSF catalyst for the partial oxidation of methane into syngas and found that the activity and selectivity of core–shell catalyst was higher than that of the Fe2O3:LSF composite catalyst. Haghighiet al. [38] synthesized a Ni-Co/Al2O3-MgO catalyst, and a strong interaction force was formed between the metal and the carrier due to high-temperature calcination.This interaction force between the bimetals transformed the structure of the catalyst into a stable spinel skeleton structure, leading to excellent performance in the reaction process; furthermore, the authors calculated the intermediate products in the reaction process and found that the formation of Ni-Co bimetals provided better carbon deposition resistance compared to the single Ni site.Kawiet al.[39]designed Ni@Ni phyllosi-replicate@SiO2core–shell hollow spheres by hydrothermal method and reduction method and found that Ni@NiPhy@SiO2catalyst showed better carbon deposition resistance than Ni@NiPhy because of the existence of SiO2shell. In our previous research [12], a series of Ni-Phyllosilicate@Ce0.8M0.2O2-δ(M = Fe, Co, Ni) redox catalysts with core–shell structure were synthesized and used for the CL-DRM process at 620 °C. It was found that transition metal-doped Ni-phyllosilicate@CeO2-δdistinctly improved its oxygen supply capacity. The outermost layer Ce0.8Fe0.2O2-δexhibited excellent structural stability in CH4or CO2atmosphere, which prevented the sintering and deactivation of metallic Ni particles. However,the effect of oxygen vacancy concentration in core–shell structure on catalyst activity was not considered.The above research shows that the core–shell structure is a practical way to promote the metal-carrier interaction effect and can be used to manufacture high-activity Ni-based catalysts for DRM.
Our work involves the synthesis of a new catalyst with sandwich core–shell structure for CL-DRM. SiO2spheres as the core were prepared by ammonia evaporation method, and the shell layer was CeO2. The sandwich layer was composed of Ni-M(M = Fe, Co, Mn, Cu, Zr) nanoparticle anchoring between core and shell.The outermost thin shell layer exhibited high oxygen storage and excellent redox performance. The abundant oxygen vacancies were regulated by Ni and transition metal(Fe,Co,Mn,Cu,Zr)alloying to improve the resistance towards carbon deposition.The synergistic effect of bimetal enhanced the dispersion and density of the surface-active sites.The high CH4conversion and CO selectivity of CL-DRM were achieved by controlling the component ratio to regulate the structure of the Ni-M/SiO2@CeO2(M = Fe, Co, Cu,Mn, Zr) core–shell catalyst and the interaction between Ni and CeO2. Our research provides a new sight for design redox catalyst in chemical looping and related field.
2. Materials and Methods
2.1. Catalyst synthesis
2 g each of Ni-M/SiO2(M = Fe, Co, Mn, Cu, Zr) was prepared by the ammonia evaporation method. SiO2nanospheres were produced by modified Stöber method [40]. During the preparation,12 ml ethyl orthosilicate and 420 ml ethanol was mixed to create a solution.Next,the solution was added by 12.6 ml of 25%ammonia and 75 ml water,and stirred at room temperature for 24 h until complete hydrolysis occurred. Then, a specific amount of nickel nitrate and cobalt nitrate (as well as ferric nitrate, copper nitrate,manganese nitrate) was added by titration. For the obtained solution,the pH was adjusted to 7 and stirred under a 70°C water bath for 4 h.After complete precipitation,the precipitate obtained from centrifugation was washed with ethanol/water five times.The prepared sample was then filtered and dried overnight at 80°C.Ni-M/SiO2(M=Fe,Co,Mn,Cu,Zr)was obtained by calcining the samples at 700 °C for 4 h, followed by grinding at room temperature.
Ni-M/SiO2@CeO2(M = Fe, Co, Mn, Cu, Zr) sandwich-like core–shell catalysts were prepared using the impregnation method according to our previous study[12].The whole catalyst synthesis process is shown in Fig. 1.
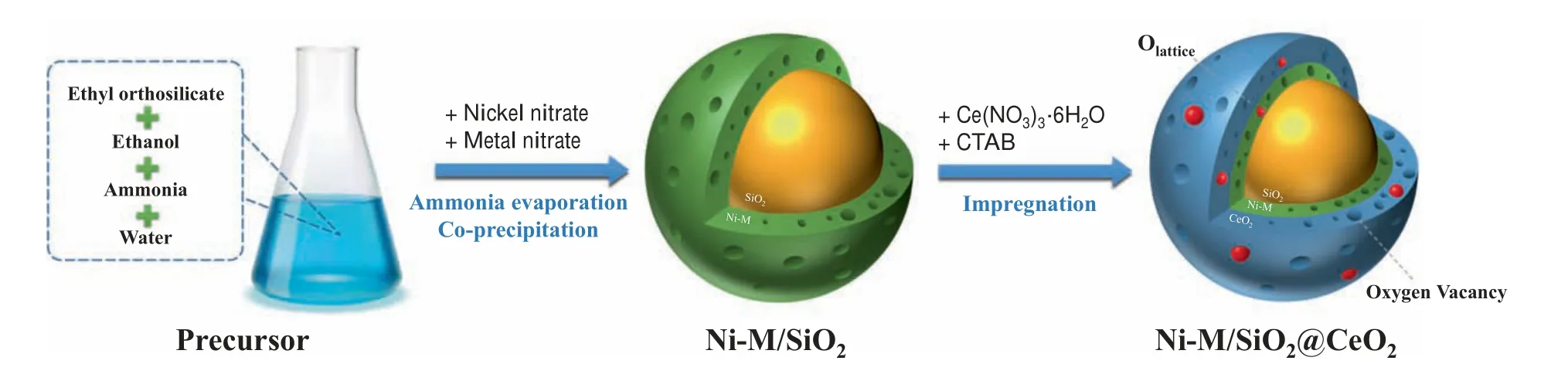
Fig. 1. Synthesis path of Ni-M/SiO2@CeO2 (M = Fe, Co, Mn, Cu, Zr) sandwich core–shell catalyst.

Fig. 2. HRTEM results of Ni-Co/SiO2 catalyst.
2.2. Catalyst characterization
The Bruner-Emmett-Teller (BET) surface areas were detected with N2adsorption (ASAP-2010, Quantachrome, America). The 150 mg sample was sequentially introduced to vacuum-drying at 120°C for 3 h,nitrogen desorption at 300°C,and nitrogen adsorption at liquid nitrogen at–196°C.According to the BET calculation,the test data were processed to obtain the corresponding specific surface area.
The phase structure and lattice size of the catalyst were determined by D/MAX-Rc X-ray diffraction (XRD) under conditions of 0.15406 nm,30 mA tube current and 40 kV tube voltage.The samples were scanned at a scanning rate of 10(°)∙min-1in the range of 20°–80°.
High resolution transmission electron microscopy (HRTEM, FEI Tecnai G2 F30, America) with acceleration voltage of 300 kV were applied for the observation of the morphology of catalysts,and the mapping surface scan was carried out to analyze the distribution of sample elements. The powder sample was ultrasonically suspended in ethanol for 10 min to prepare the specimen. A drop of suspension was deposited on the copper grid supported by a thin carbon film. Then the suspension was dried in air to obtain the specimen. Because the sample contains magnetic Ni, it cannot be tested directly, so the sample was sandwiched in the middle by two layers of copper mesh.
The surface properties of the catalyst were determined by Raman spectrometer (Thermo Fisher DXRxi, Thermo Fisher Scientific Co., Ltd., America). The used laser wavelength, laser power,power, spectral range and spectral resolution was 532 nm, 0.4 mW, 3 mW, 100–1600 cm-1and 5 cm-1respectively, while the exposure time was 0.5 s.
The H2-Temperature programmed reduction (H2-TPR) experiment was carried out on the thermal conductivity detector of the ChemBET Pulsar adsorption instrument(Quantachrome, America).A 50 mg sample loaded in a U-shaped quartz tube was pretreated at 200°C for 1 h under N2atmosphere(30 ml∙min-1).After cooling to room temperature,the sample was heated from room temperature to 900°C at 10°C∙min-1in flow of 10% (vol)H2/N2mixed gas(75 ml∙min-1).
2.3. Reaction performance test
CH4-TPR experiment was conducted to evaluate the methane reactivity over the catalyst. The 300 mg catalyst particles were weighed by an electronic balance, and CH4-TPR was performed on a self-built experimental platform (CATLAB, Heaton, UK); the gas composition was detected. In the reactor, the sample was pre-treated with high-purity Ar (50 ml∙min-1) as an inert protective gas at a constant temperature of 300 °C for 1 h, and then 5%(vol) CH4/Ar (25 ml∙min-1) was introduced to the reactor at temperatures ranged from 300 °C to 800 °C at a heating rate of 10 °C∙min-1. Reaction exhaust gas was continuously analyzed and tested in real time by an online mass spectrometer (QGA,Hilden, UK), and the concentration signals of CH4, H2, CO and CO2were recorded.
CO2temperature programmed oxidation (CO2-TPO) test was also performed to determine the adsorption and reactivity of carbon dioxide at different temperatures of the catalyst in the reduced state.The catalyst was reduced at a constant temperature of 650°C for 10 min.After cooling to room temperature under the protection of Ar atmosphere,and then the inlet valve was switched to 5%(volume) CO2/Ar (three-necked flask mixed with water vapor,50 ml∙min-1). The temperature of reactor was heated from room temperature to 800 °C at 10 °C∙min-1. Reaction exhaust gas was analyzed and tested in real time by an online mass spectrometer,and the concentration signals of CH4, CO, CO2and H2were recorded.
Redox cycle stability test was performed using the same reactor system. The experiment was carried out using 100 mg samples at 0.1 MPa and 600°C.The reactor was rinsed with Ar(50 ml∙min-1)before the test. The sample was isothermally reduced at 5% (vol)CH4/Ar and isothermally oxidized at 5% (vol) CO2/Ar at a flow rate of 50 ml∙min-1. The reduction and oxidation were carried out for 10 min each cycle, and the reactor was purged with Ar for 10 min between each cycle to avoid the mixing of reduction and oxidation gases. The gas composition at outlet of reactor was analyzed by a mass spectrometer(LC-D200M PRO,TILON,Japan).Use a gas chromatograph(7890B,Agilent Technologies Co.Ltd.,America)with a gas sampling bag to periodically analyze the gas composition offline to check the accuracy of the analysis.
Through the calculation of CH4conversion, CO selectivity and hydrogen-carbon ratio, the performance of the reaction catalyst can be more intuitive. The specific calculation formula was as follows:
wheren1is the amount of methane consumed,n2is the amount of methane introduced,n3is the carbon monoxide production,n4is the total production of carbon monoxide and carbon dioxide,n5is the hydrogen production.
3. Results and Discussion
3.1. Synthesis of core–shell redox catalyst
HRTEM was performed on freshly prepared catalyst samples to analyze their structure.From Fig.2,it can be observed that the uniform silica nanospheres synthesized by the Stöber method [40],had a diameter of 70 nm. During the followed evaporation of ammonia, nickel and cobalt nitrates were precipitated on the surface of SiO2nanospheres to form layered silicate species creating a thin fibrous layer. It is clear from the energy dispersive spectrometer(EDS)mapping of the catalyst precursor(Fig.2(c)and(d))that the nanoparticles of Ni and Co are uniformly dispersed on the silicon spheres. The HRTEM image and EDS mapping of Ni-Co/phyllosilicate catalyst demonstrate that the core–shell structure is composed of a silicon core, and the nanoparticles of Ni and Co are attached on the surface of the silica sphere.
The impregnation method was used to prepare Ni-M/SiO2@CeO2(M=Fe,Co,Cu,Mn,Zr)on the basis of Ni-M loaded silicate spheres.Fig. 3(c)–(f)shows the EDS mapping of the catalysts of Ni-Co/SiO2@CeO2. The results of elements distribution clearly indicates that the structure of catalysts is the sandwich structure with an inert silicon core and a thin CeO2shell (~5 nm) where the Ni-Co nanoparticles are evenly distributed on the silicon core surface.

Fig. 3. HRTEM images of Ni-Co/SiO2@CeO2 catalysts.
3.2. Synergy effect of ni-transition metals in redox catalyst
3.2.1. Phase composition
The phase composition and reducibility of core–shell Ni-M/SiO2@CeO2(M = Fe, Co, Cu, Mn, Zr) catalysts are shown in Fig. 4.As shown in the XRD patterns (Fig. 4(a)), the sharp diffraction peaks evidenced high crystallinity. The six main diffraction peaks observed at 28.6°, 33.1°, 47.5°, 56.4°, 69.6° and 76.7° correspond to the cubic fluorite phase of ceria [12,41]. An obvious blue shift of main characteristic peak of ceria was observed for Co, Mn and Cu doped catalysts,revealing the transition metal cations incorporated into ceria lattice and formed Ce-M-O solid solution. The formation of solid solution will enhance lattice distortion and create abundant oxygen vacancies [42]. A weak peak is appeared at 37.2°, 43.2° and 79.4°, indicating the existence of nickel oxide. In addition, the characteristic peaks of the phyllosilicate were also detected.Generally,XRD patterns show that the poorly crystallized nickel phyllosilicate phase coexists with nickel oxide and silica.As shown in Fig. 4(a), the unreduced samples are presence of corresponding metallic oxide crystal phase (Co3O4, Fe3O4, CuO,MnO, ZrO2) and some reduced nickel oxide species. However, no diffraction peaks of nickel metal were detected on the pretreated catalysts. It might be ascribed to the highly dispersed nature of metallic Ni on the surface of the silica spheres or the low content of nickel.
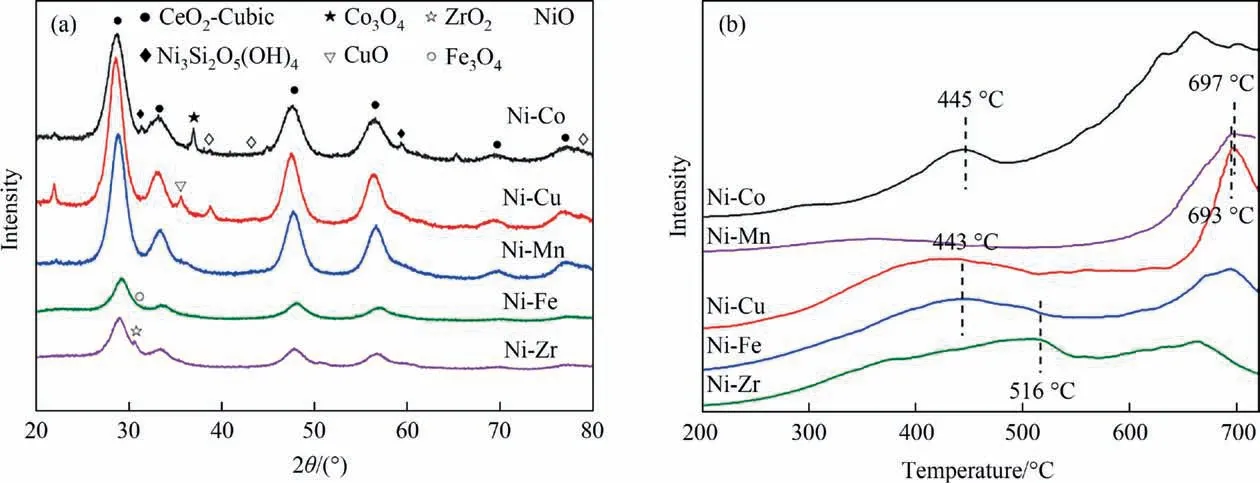
Fig. 4. (a) XRD patterns and (b) H2-TPR patterns of fresh Ni-M/SiO2@CeO2 (M = Fe, Co, Cu, Mn, Zr) catalysts.
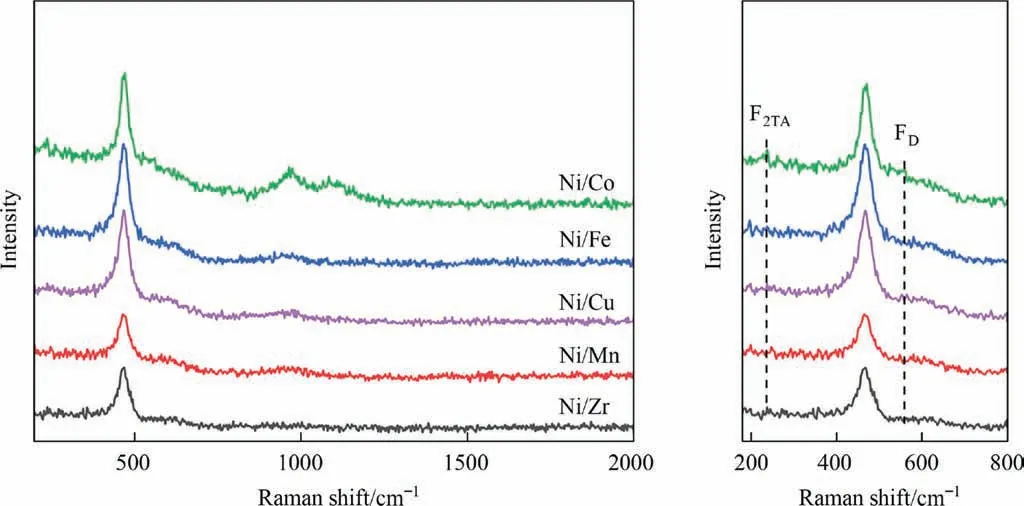
Fig. 5. Raman spectra of Ni-M/SiO2@CeO2 (M = Fe, Co, Cu, Mn, Zr) redox catalysts.
The impact of transition metal modification on catalysts reducibility was studied in H2-TPR experiments[43].Fig.4(b)indicates that the reduction curves of Ni-M/SiO2@CeO2(M=Fe,Co,Cu,Mn,Zr)catalysts show two broad reduction peaks derived from the overlap of the reduction of Ni2+, transition metal oxides and ceria at temperatures ranged from 300 to 700 °C. The reduction of Ni2+in the Ni-M/phyllosilicate was detected between 450 °C and 750°C[39].The results indicate that the silicate species are formed in Ni-M@SiO2catalysts due to roasting in the preparation process and a strong interaction between SiO2core, Ni-M sandwich layer and CeO2shell layer, further confirming the morphology structure of the catalyst.The reduction peaks of Ni-M/SiO2@CeO2(M=Fe,Co,Cu) catalysts obviously shift to the lower temperatures due to enhanced oxygen mobility, indicating that the strong interaction among transition metal oxides and ceria. Some transition metal oxides might incorporate into ceria during the calcination. This result implies the core–shell and sandwich structure of the catalysts facilitates the alloying of nickel and transition metals during the reduction process, which promotes the reducibility of the catalysts [12,44].
3.2.2. Oxygen vacancy concentration
The oxygen vacancy of catalyst is conducive to both adsorption of reactants and dissociation of products [44]. The surface lattice oxygen of ceria will be reduced in a reducing atmosphere,creating abundant oxygen vacancies on the surface.Subsequently,the oxygen vacancies in CeO2are refilled through the processwith gaseous oxygen[16,45]. Besides, oxygen vacancies in ceria-based catalysts will help to anchor active species to create active site for catalytic reaction. The existence of suitable amount of oxygen vacancies will enhance oxygen mobility and OSC. The influence of transition metal doping on the oxygen vacancy concentration was studied using Raman spectroscopy for Ni-M/SiO2@CeO2(M = Fe, Co, Cu,Mn, Zr) redox catalysts.
The Raman spectroscopy curves in Fig.5 shows that the peak at 460 cm-1is owing to the F2gvibration mode in ceria fluorite structure, and the peaks at 262 and 598 cm-1are ascribed to the F2ATvibration mode and defection-induced FDvibration mode, respectively. The ratio of the peak integrated area of FDto F2grepresents the relative concentration of oxygen vacancy on the ceria surface[46]. The oxygen vacancy concentration of the Ni-M/SiO2@CeO2(M = Fe, Co, Cu, Mn, Zr) catalysts follows an order of Co > Cu > Fe > Mn > Zr. Among them, Ni-Co/SiO2@CeO2has the highest concentration of oxygen vacancy, which is most likely due to the incorporation of Ni and Co cations into ceria lattice and the interaction between Ni and Co oxides [20]. These results further confirmed the strong interaction between Ni-Co alloy and ceria obtained in H2-TPR.
3.2.3. CH4/CO2 activation
Based on the low surface oxygen vacancy concentration and poor reduction performance of Ni-Mn/SiO2@CeO2and Ni-Zr/SiO2@CeO2, both of them are not studied in subsequent experiments. Fig. 6(a)–(c) indicates the change of gas composition and methane conversion of a series Ni-M/SiO2@CeO2(M = Fe, Co, Cu)catalysts in CH4-TPR experiments. In CH4-TPR, the gas–solid reaction between CH4and catalyst is dominated by a selective oxidative of CH4into H2and CO, and the full oxidation of CH4is depressed [47]. When the lattice oxygen is consumed, methane will split into H2with the formation of deposited carbon [48].Fig.6(a)–(c)shows that the reactivity of the three catalysts is very poor when the temperature is lower than 600 °C. The onset temperatures of the three catalysts are in order of Ni-Co/SiO2@CeO2(600 °C) < Ni-Fe/SiO2@CeO2(640 °C) < Ni-Cu/SiO2@CeO2(700 °C).The time from the beginning of the reaction to the time of the highest methane conversion is shorter for Ni-Co/SiO2@CeO2, indicating high mobility of selective lattice oxygen in the catalyst.It suggests that compared to Fe and Cu,the addition of Co on catalyst has better methane conversion performance and lower onset temperature of methane activation.
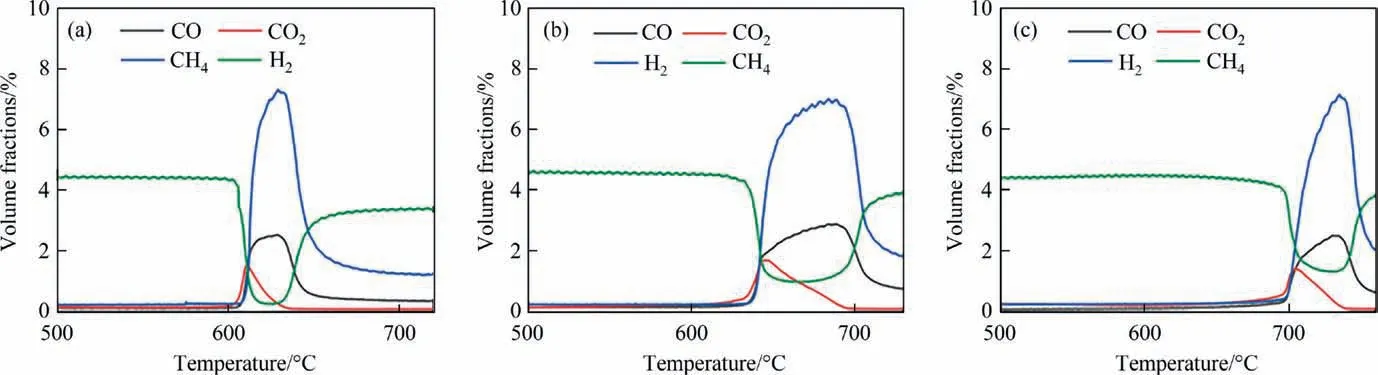
Fig. 6. CH4-TPR profiles for Ni-M/SiO2@CeO2 redox catalysts (M = (a) Co, (b) Fe, (c) Cu).
The CO2-TPO experiment was conducted to evaluate the reactivity of CO2over the reduced catalysts. As shown in the Fig. 7,the reduced Ni-M/SiO2@CeO2(M = Fe, Co, Cu) could split CO2into CO and regenerate lattice oxygen. The onset temperature of CO2-TPO over the reduced catalysts are 590, 610 and 650 °C for Co, Fe and Cu doped ones, which is consistent with order of onset temperature of CH4-TPR. Ni-Co/SiO2@CeO2shows a strong CO peak since more lattice oxygen was removed from the catalyst.
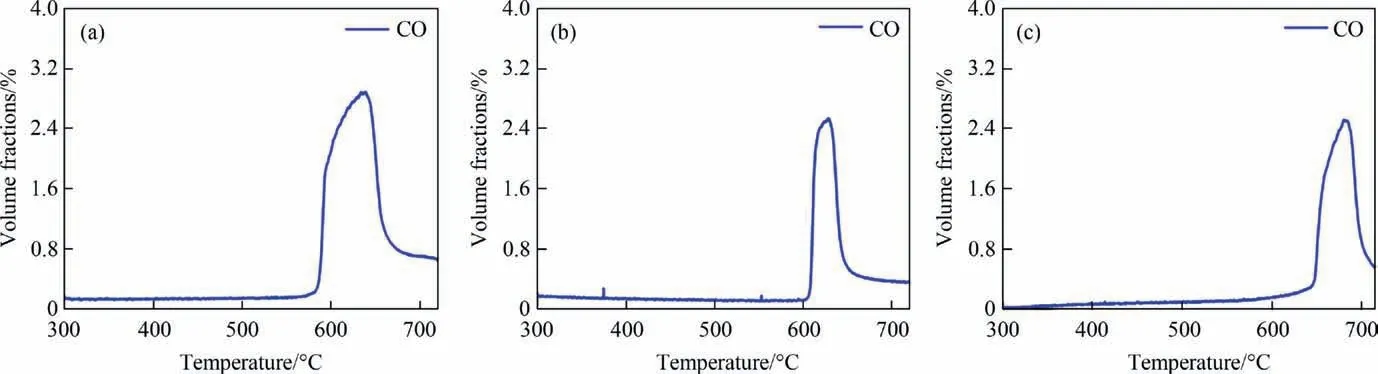
Fig. 7. CO2-TPO profiles for Ni-M/SiO2@CeO2 redox catalysts (M = (a) Co, (b) Fe, (c) Cu).
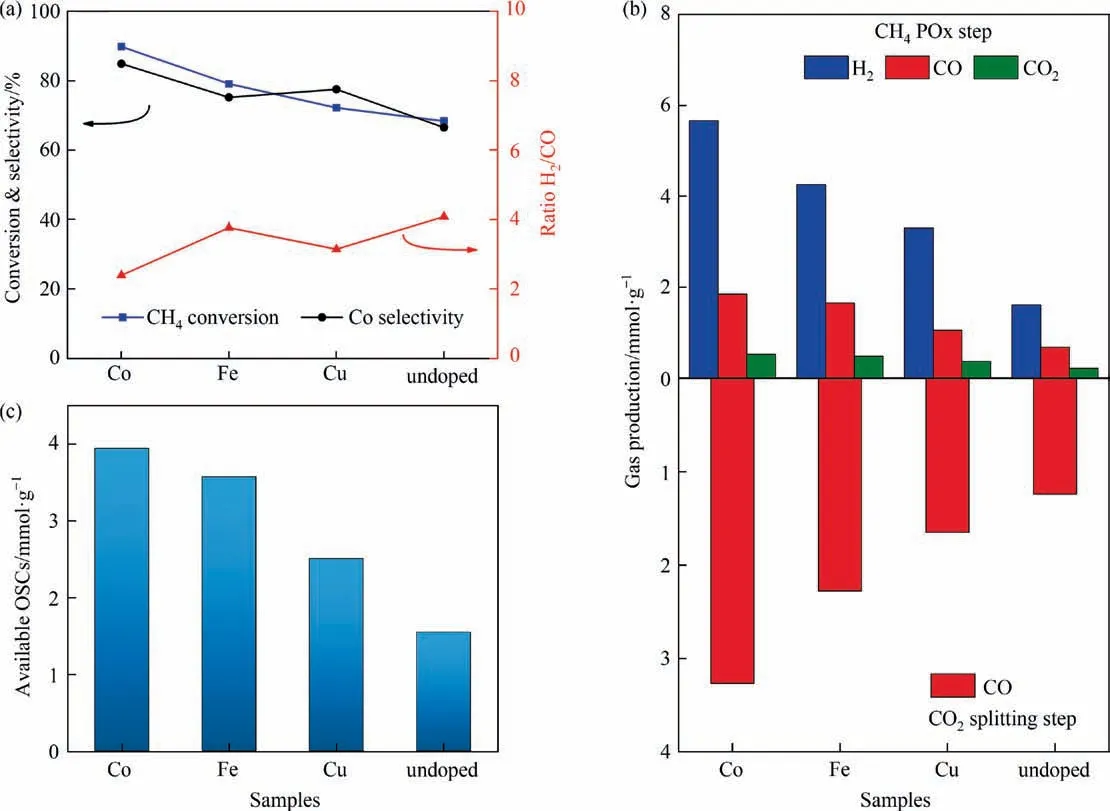
Fig. 8. (a) The conversion performance in CH4 partial oxidation (POx) steps at 615 °C, (b) gaseous product content and (c) available oxygen storage during CH4-POx/CO2-splitting steps over Ni/SiO2@CeO2 and Ni-M/SiO2@CeO2 (M = Fe, Co, Cu).
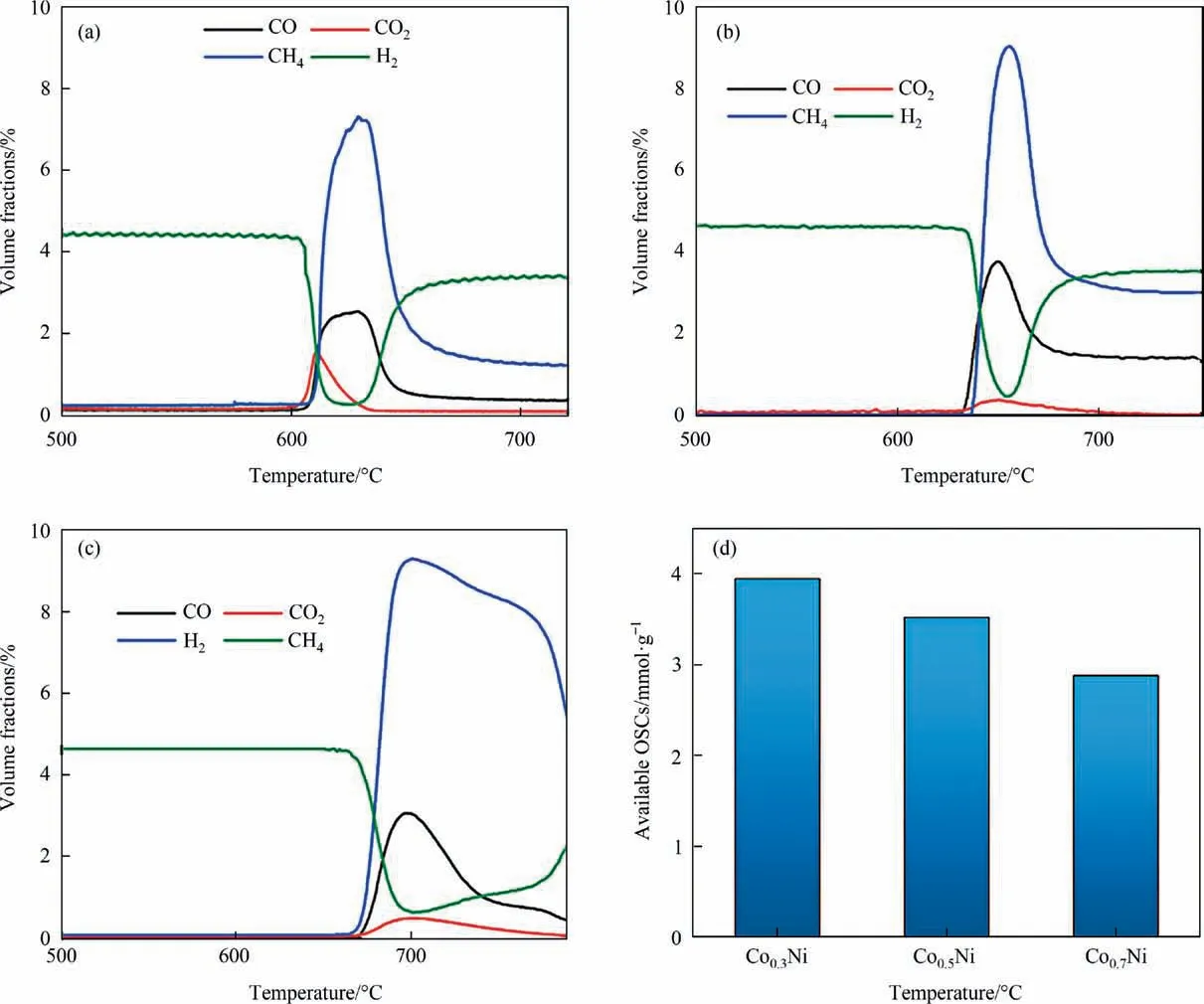
Fig.9. CH4-TPR curves of(a)Ni-Co0.3/SiO2@CeO2,(b)Ni-Co0.5/SiO2@CeO2,and(c)Ni-Co0.7/SiO2@CeO2 catalysts;(d)the available oxygen storage of catalysts during CH4 POx and CO2 splitting.
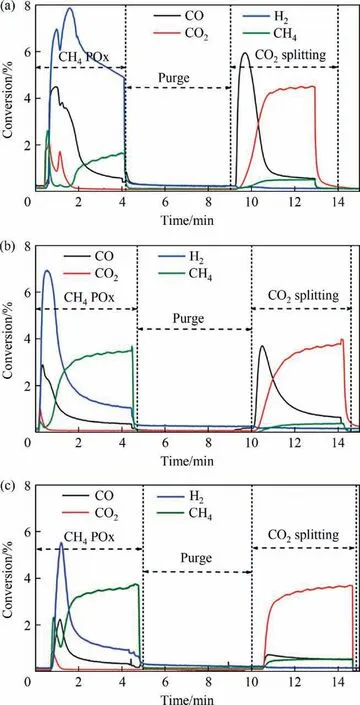
Fig.10. The gas composition in CH4 POx and CO2 splitting reactions at 615°C over(a) Ni-Co0.3/SiO2@CeO2, (b) Ni-Co0.5/SiO2@CeO2, and (c) Ni-Co0.7/SiO2@CeO2 catalysts.
Fig.8(a)shows the performance of CH4isothermal reaction over Ni-M/SiO2@CeO2(M = Fe, Co, Cu) catalysts at 615 °C. Among undoped and three doped catalysts, Ni-Co/SiO2@CeO2presents highest CH4conversion and CO selectivity following with a lowest H2/CO molar ratio(2.38)close to idea ratio of 2.It suggests that the catalyst enabled to selectively oxidize CH4into syngas using lattice oxygen, and the cracking and fully oxidation of CH4were depressed. In contrast, Ni/SiO2@CeO2presents a lowest CH4conversion with a highest H2/CO molar ratio (4.08) due to weak oxygen donation ability and lower catalytic activity.Correspondingly, both the product amounts from CH4partial oxidation and CO2splitting steps follow an order of Co>Fe>Cu>undoped for Ni-M/SiO2@CeO2catalyst.Fig.8(c)confirmed that Ni-Co/SiO2@CeO2demonstrated a highest available OSC throughout redox steps, which is consistent with high OSC and oxygen mobility mentioned in Section 3.2.2.
On the basis of Ni-Co/SiO2@CeO2, the molar ratio of Co/Ni was optimizedviaCH4-TRP experiments, as shown in Fig. 9. Fig. 9(a)–(c) indicates that the onset temperature follows an order of Ni-Co0.7/SiO2@CeO2(678 °C) > Ni-Co0.5/SiO2@CeO2(642 °C) > Ni-C o0.3/SiO2@CeO2(606 °C). CH4-TPR profiles shows that the increase of Co amount in Ni-Co0.7/SiO2@CeO2shifted the methane reaction to higher temperature and led to methane cracking. The available OSC in Fig. 9(d) shows that Ni-Co0.3(3.95 mmol∙g-1) > Ni-Co0.5(3.52 mmol∙g-1) > Ni-Co0.7(2.88 mmol∙g-1). Obviously, the lowtemperature reactivity was depressed and the high-temperature cracking of methane was enhanced for the Ni-Co/SiO2@CeO2with higher Co/Ni molar ratios. This is mainly due to that high Co/Ni ratio can result in oxidation deactivation of metals in the catalyst while the drop of Co/Ni ratio increases the methane activation but leads to carbon deposition on the catalyst surface [49]. Therefore,the catalyst with the highest CH4conversion and CO selectivity was obtained by controlling Co/Ni molar ratio of 0.3.
3.3. Cycling stability in successive CH4-POx/CO2-splitting cycles
The successive CH4-POx/CO2-splitting cycles were carried out at 615°C using Ni-Co/SiO2@CeO2catalysts.The influence of the Co/Ni ratio on the product gas components in redox reactions over Ni-Co/SiO2@CeO2catalysts was investigated. The gas composition in the third redox cycle over Ni-Co/SiO2@CeO2catalysts with Co/Ni molar ratios of 0.3, 0.5 and 0.7 are shown in Fig. 10.
It is obvious that three catalysts show distinct redox kinetics.Compared to other two catalysts, Ni-Co0.3/SiO2@CeO2exhibited strongest redox reactivity, achieving nearly 100% conversion for both CH4partial oxidation and CO2splitting reactions in the beginning. For the CH4POx step, sharp peaks of CO2, CO and H2were observed in the beginning of the reaction due to the high mobility of O2–.Besides,the H2/CO molar ratio of syngas close to 2 was produced over Co-enriched catalysts since Co species are only active to methane cracking at higher temperature (>650 °C). Methane tended to crack into hydrogen and deposited carbon in the final stage of the reaction over Ni enriched catalyst due to its lowtemperature activation performance [47]. Some highly active lattice oxygen resulted in fully combustion of methane, which depressed CO selectivity. In CO2-splitting step, CO peaks were observed in CO2splitting reactions over all catalysts except Ni-Co0.7/SiO2@CeO2catalysts. CO2was nearly completely converted into CO in the beginning of the reaction. This weak CO2splitting activity over Ni-Co0.7/SiO2@CeO2might imply that CO2cannot regenerate lattice oxygen consumed in CH4POx. It is worth to notice that both CO2splitting/oxygen regeneration rates were decreased with Co doping. A suitable Co/Ni molar ratio in the Ni-Co/SiO2@CeO2modulate oxygen donation/regeneration ability for efficient conversion of CH4and CO2in a chemical looping mode.
The redox stability of Ni-Co0.3/SiO2@CeO2catalyst were evaluated in the successive CH4-POx/CO2-splitting redox cycles, as shown in Fig. 11. The CH4conversion and CO selectivity slightly increases in the first five cycles,which is defined as a stabilization stage for the catalyst. Generally, oxygen carrier or redox catalyst will experience a stabilization treatment following with the selfadaptive process of the materials in aspects of phase composition and microstructure in the beginning redox cycles. Thereafter, the CH4conversion tends to stabilize during the subsequent redox cycles.A CO2selectivity around 15%was observed during the redox cycles because of active surface oxygen[47,50].It also implies that the catalyst showed high oxygen regenerability and oxygen diffusion ability, thus creating very active surface lattice oxygen in redox atmospheres without strong oxidants.According to the relative constant production in redox cycles, the reduced catalyst was well-regeneratedviaCO2splitting and the deposited carbon from methane cracking was completed gasified to produce COviathe carbon-CO2gasification reaction [51]. In general, Ni-Co0.3/SiO2@CeO2catalyst showed high redox stability for the continuous generation of syngas and CO in 20 redox cycles, achieving 87% CH4conversion, 83% CO selectivity, 2.28 H2/CO molar ratio in average.
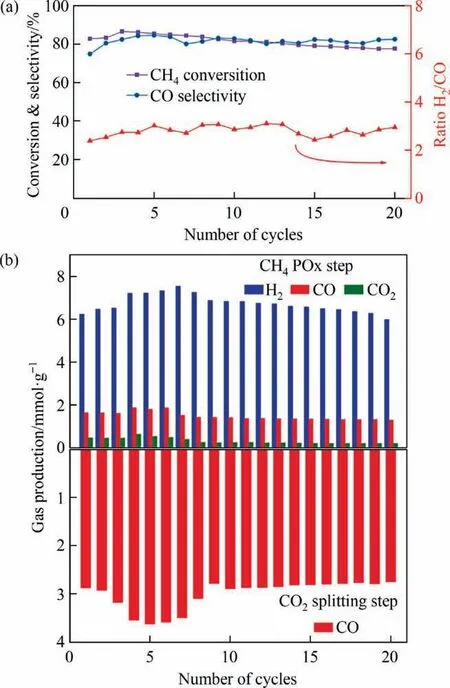
Fig. 11. (a) CH4 POx performance in the successive CH4/CO2 redox cycles over Ni-Co0.3/SiO2@CeO2 catalyst at 615 °C. (b) Amounts of H2, CO, and CO2 gaseous products generated in the redox cycles over Ni-Co0.3/SiO2@CeO2 catalyst.
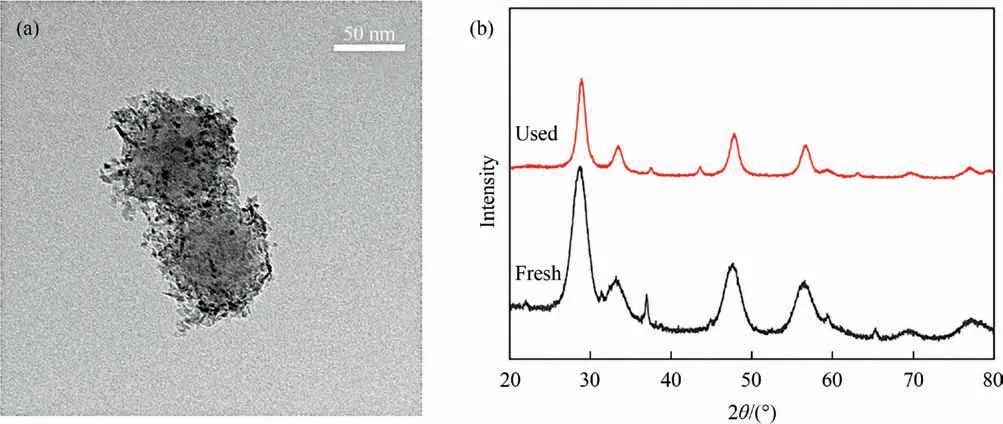
Fig. 12. (a) HRTEM images of post-cycle Ni-Co0.3/SiO2@CeO2 catalysts and (b) XRD patterns for fresh and used Ni-Co0.3/SiO2@CeO2 obtained from 20 redox cycles.
Fig.12 illustrates the HRTEM and XRD results of Ni-Co0.3/SiO2@CeO2catalyst obtained after 20 redox cycles. The catalyst maintained a stable core–shell structure even after 20 CH4/CO2redox cycles. No structural collapse was observed and the size of the spherical structure remained consistent with that of the freshly prepared samples. The surface of SiO2nanosphere is still tightly surrounded by nanoparticles of ceria, Ni-Co metal/metal oxides.More importantly, no carbon deposition was found on the surface of the catalyst.The core–shell structure ensured the metallic nickel particles were stably fixed between the spherical core and the thin shell of ceria [12]. This unique structure enhances the oxygen delivering/acquisition ability of catalyst during CL-DRM process,inhibiting deactivation of the catalyst and heavy carbon deposition throughout redox cycles[52,53].Fig.12(b)displays the XRD results for the fresh and recovered Ni-Co0.3/SiO2@CeO2catalysts. Both samples exhibit similar XRD patterns but with lower peak intensity[54]. No obvious shift of main characteristic peaks was observed.This suggests that ceria in the catalyst maintained high structural stability in reversible oxygen donation/regeneration cycles. The diffraction peak near 37° was attributed to the formation of Ni-Co alloy. All diffraction peaks were weakened and shrunk after long-term redox cycles due to enhanced dispersion and crystallization. Both the results of redox activity and structural evolution of the catalyst confirmed that Ni-Co0.3/SiO2@CeO2demonstrated high reactivity and structural stability in the successive CH4/CO2redox cycles for the generation of syngas and CO.
4. Conclusions
A series of core–shell structured Ni-M/SiO2@CeO2redox catalysts(M=Fe,Co,Cu,Mn and Zr)were synthesized and investigated in a fixed-bed reactor for CL-DRM. The catalysts are composed of spherical SiO2core with a diameter of 70 nm and CeO2shell, and the highly dispersed Ni alloy nanoparticles are the interlayer between core and shell. The interaction between CeO2and Ni or transition metal oxides is favorable for the formation of ceriabased solid solution and creation oxygen vacancies, thus promoting dispersion of Ni or Ni-M alloys. The OSC of Ni-M/SiO2@CeO2catalyst follows an order of Co>Cu>Fe>Zr>Mn and the oxygen vacancy concentration follows an order of Co > Cu > Fe > Mn > Zr.CH4-TPR and CO2-TPO indicates that the catalysts with higher OSC and concentration of oxygen vacancy exhibited lower CH4/CO2activation temperatures. Ni-Co/SiO2@CeO2showed highest redox activity and the rationalization of Ni/Co proportion further enhance available OSC to increase CH4/CO2conversion. In isothermal CH4/CO2redox, Ni-Co0.3/SiO2@CeO2enabled to partial oxidize methane to syngas with a CH4conversion of 87%and CO selectivity of 83%, and then was regeneratedviaCO2splitting. The catalyst revealed superior activity and structural stability in the successive redox cycles at 615 °C for the production of syngas with a molar ratio close to 2. The proposed catalyst shows great potential for the efficient conversion of CH4and CO2in a redox mode at moderate temperatures.It also offers an insight into design redox catalyst for chemical looping and related fields.
Data Availability
Data will be made available on request.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52066007, 22279048), Yunnan Major Scientific and Technological Projects (202202AG050017), and the Applied Basic Research Program of Yunnan Province (202101AT070076).
Nomenclature
n1amount of methane consumed
n2amount of methane introduced
n3carbon monoxide production
n4total production of carbon monoxide and carbon dioxide
n5hydrogen production
 Chinese Journal of Chemical Engineering2023年11期
Chinese Journal of Chemical Engineering2023年11期
- Chinese Journal of Chemical Engineering的其它文章
- Effects of the original state of sodium-based additives on microstructure,surface characteristics and filtration performance of SiC membranes
- Comprehensive analysis on the economy and energy demand of pressure-swing distillation and pervaporation for separating waste liquid containing multiple components
- Esterification of acetic acid with isobutanol catalyzed by ionic liquid n-sulfopropyl-3-methylpyridinium trifluoromethanesulfonate:Experimental and kinetic study
- Numerical investigation of film forming characteristics and mass transfer enhancement in horizontal polycondensation kettle
- COF-derived Co nanoparticles@N-doped carbon electrocatalysts for highperformance Zn-air batteries
- A potential-responsive ion-pump system based on nickel hexacyanoferrate film for selective extraction of cesium ions