
Fabrication of highly dispersed carbon doped Cu-based oxides as superior selective catalytic oxidation of ammonia catalysts via employing citric acid-modified carbon nanotubes doping CuAl-LDHs
2024-01-13 04:53FengrongLiXuezhenLiuZhengYiZhaoXiaAnYaliDuXuWu
Fengrong Li, Xuezhen Liu, ZhengYi Zhao, Xia An, Yali Du, Xu Wu,
1 College of Chemistry, Taiyuan University of Technology, Taiyuan 030024, China
2 College of Chemical Engineering and Technology, Taiyuan University of Technology, Taiyuan 030024, China
3 College of Chemistry and Chemical Engineering, Jinzhong University, Jinzhong 030619, China
Keywords: Selective catalytic oxidation of ammonia Layered-double hydroxides Cu-based oxides CNTs Citric acid-modified
ABSTRACT In this work,the CuAl-LDO/c-CNTs catalyst was fabricated via in situ oriented assembly of layered-double hydroxides(LDHs)and citric acid-modified carbon nanotubes(c-CNTs)followed by annealing treatment,and evaluated in the selective catalytic oxidation(SCO)of NH3 to N2.The CuAl-LDO/c-CNTs catalyst presented better catalytic performance (98% NH3 conversion with nearly 90% N2 selectivity at 513 K) than other catalysts, such as CuAlOx/CNTs, CuAlOx/c-CNTs and CuAl-LDO/CNTs. Multiple characterizations were utilized to analyze the difference of physicochemical properties among four catalysts. XRD, TEM and XPS analyses manifested that CuO and Cu2O nanoparticles dispersed well on the surface of the CuAl-LDO/c-CNTs catalyst. Compared with other catalysts,larger specific surface area and better dispersion of CuAl-LDO/c-CNTs catalyst were conducive to the exposure of more active sites,thus improving the redox capacity of the active site and NH3 adsorption capacity. In-situ DRIFTS results revealed that the internal selective catalytic reduction (iSCR) mechanism was found over CuAl-LDO/c-CNTs catalyst.
1. Introduction
Nitrogen oxides(NOx), emitted from stationary sources such as coal-fired power plants and industrial boilers, severely jeopardize the ecological environment and human health [1–3]. Therefore, it is extremely urgent to control NOxemission from these sources.Compared with other de-NOxtechnologies, selective catalytic reduction (SCR) of NOxwith NH3(NH3-SCR) is one of the most effective ways to reduce NOxemissions [4–6]. In the SCR process,ammonia (NH3) was used as reducing agent to convert NOxinto N2and H2O [2,6,7].However,increasing the NH3/NO ratio inevitably leaded to NH3slip for the SCR process[5,8].Excessive NH3has been released into the atmosphere which has caused serious harm to humans and the ecological environment [8–10]. Selective catalytic oxidation of ammonia (NH3-SCO) is supposed as the most potential deamination methods to eliminate the slip ammonia[9–12]. Currently, it was significant to design a suitable catalyst about NH3-SCO [9,13].
In the last several decades, many transition metal (Cu, Mn, Co,Fe)oxides have been intensively applied as ammonia slip catalysts(ASCs).According to II’chenko research,CuO catalyst was reported as one of the most active and efficient catalysts for NH3oxidation[14].In later studies on pre-oxidized polycrystalline copper foils,it was proved that CuO phase was the active phase of NH3-SCO[15].However, the foremost issue was poor N2selectivity in the NH3-SCO reaction for CuO catalyst. Further studies concerning NH3-SCO were carried out over the Cu-based composite oxides catalysts,i.e.Cu-Al[16–18].Lianget al.[16]studied the Cu/γ-Al2O3catalysts for NH3-SCO reaction by incipient wetness impregnation and the result displayed that the Cu (AC) catalyst presents excellent oxidation activity(T100%=623 K,>92%selectivity for N2).However,the uncoordinated activity and N2selectivity at low temperatures(<623 K), poor stability remain the main restricting factors for application. Based on this,Songet al.[19]studied the SCO properties over CuO/CNTs catalyst using CNTs as carrier. The results presented that the introduction of CNTs could balance the catalytic activity and N2selectivity of the catalyst at low temperatures to a certain extent.However,due to the defects of traditional impregnation methods, the dispersion of the active components of the above-mentioned catalyst still needs to be further improved.Therefore,how to effectively assemble the CuAlOxand the CNTs to further strengthen the balance between activity and nitrogen selectivity and improve the dispersion of the active components has attracted our attention.
The layered double oxides (LDO) catalysts derived by CuAl layered-double hydroxides (LDHs,mH2O have the advantages of large specific surface, and were widely applied in catalytic reactions [20–25]. Citric acid could be used as a blocker because of its complexation[24,26,27],whether it is possible to derive CNTs doped Cu-based oxides catalyst by modifying CuAl-LDHs with citric acid-modified CNTs raised our concerns.
In this paper, based on the excellent structural and physiochemical properties of LDHs (layered double hydroxides) and CNTs, by introducing citric acid (CA) as a blocker, a CuAl-LDH/c-CNTs catalyst was novelly fabricated by modifying CuAl-LDHs with citric acid-modified CNTs and the derived CuAl-LDO/c-CNTs were further used in NH3-SCO. Meanwhile, a strict comparison of the activity of the CuAl-LDO/c-CNTs catalysts was attempted against CuAlOx/CNTs, CuAlOx/c-CNTs and CuAl-LDO/CNTs catalysts. A series of characterizations including XRD, SEM, TEM, N2adsorption–desorption analysis,XPS,H2-TPR,NH3-TPD andin situDRIFTS were performed to establish the relationship between the catalytic performance and the physiochemical properties. Especially, the importance of active component dispersion on the surface of the catalyst was fully discussed.This work may expand a new horizon to fabricate LDO catalyst using LDHs as precursor in the field of NH3-SCO.
2. Experimental
2.1. Catalyst preparation
All chemicals were of analytical grade and were employed as received. Pristine multi-wall carbon nanotubes (MWCNTs) were purified by re-fluxing in 68% (mass) nitric acid at 393 K for 6 h.Then the purified CNTs were filtered at room temperature,washed with de-ionized water until neutral, and dried overnight at 343 K to obtain CNTs powder.
The CuAl-LDHs/c-CNTs precursor was prepared by anin situprecipitation assembly method, the mole ratio of Cu2+:Al3+= 3:1[24]. Firstly, the treated CNTs power (200 mg) was dispersed in de-ionized water to form solution A. Then, CA (200 mg) was dissolved in the solution A by sonication for 5 min to form the solution B. The mixed solution (NaOH = 0.15 mol∙L–1and Na2CO3=0.05 mol∙L–1) was slowly dripped into a round-bottomed flask within solution B while stirring vigorously to adjust the pH of solution B to 10. The nitrate mixed solution (Cu(NO3)2= 0.3 mol∙L–1,Al(NO3)3= 0.1 mol∙L–1) and the mixed solution (NaOH = 0.15 mol∙L–1and Na2CO3=0.05 mol∙L–1)was slowly dripped into above solution B. The pH value of final mixed solution was close to 10.After that, the mixed solution was hydrothermally treated at 338 K for 10 h, then the LDH slurry was filtered, washed with de-ionized water and dried overnight at 343 K,followed by annealing at 773 K for 5 h in N2atmosphere to obtain the CuAl-LDO/c-CNTs catalyst. Schematic of the synthesis strategy for CuAl-LDO/c-CNTs catalyst is displayed in Fig. 1. First, CA was grafted on the surface of modified CNTs by hydrogen bonding or chemical bonding between carboxyl groups (—COOH) and hydroxyl groups(—OH)on both CA ions and CNTs surface.Afterwards,the carboxyl groups of CA were deprotonated by adjusting the pH of the solution, and function as a coordination site to capture the metal cations. Thus, the deprotonated carboxyl groups of the CA could work as a bridge between the CNTs and metal cations, which can promote the oriental nucleation of metal cations on the CNTs surface after the addition of precipitators. Consequently, the thin LDHs werein situassembled on the CNTs surface and the CuAl-LDO/c-CNTs catalyst can be obtained after further calcination.In addition, the thermal stability of CuAl-LDHs/c-CNTs precursor was displayed in Fig.S1(Supplementary Material).The preparation method of CuAl-LDO/CNTs catalyst was similar to that of the CuAl-LDO/c-CNTs catalyst except that the mass of CA was 0 mg.
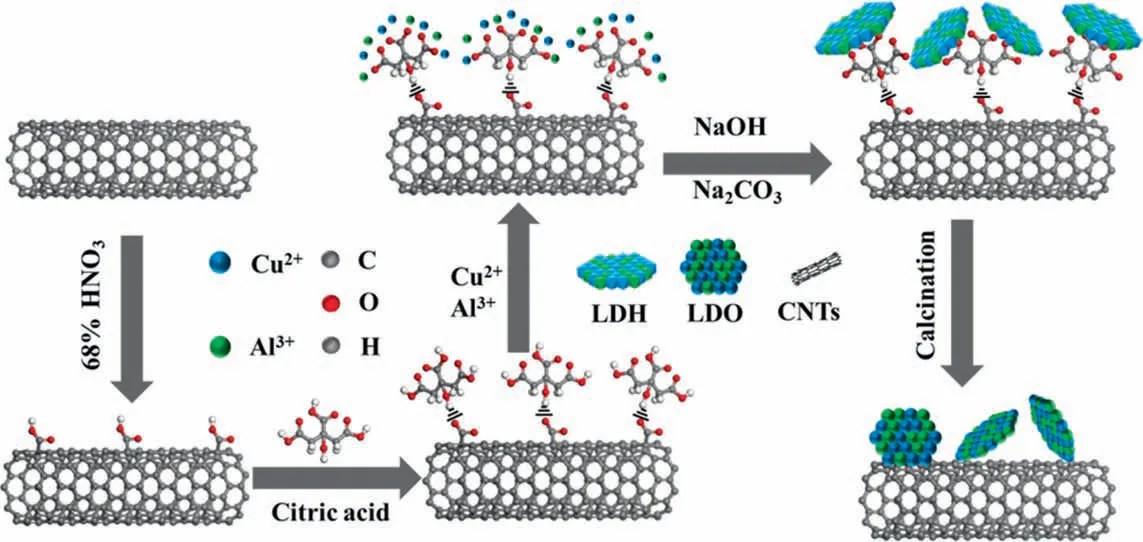
Fig. 1. Schematic of the synthesis strategy for CuAl-LDO/c-CNTs catalyst.
In addition, the 10% (mass) Cu-Al/CNTs (denoted as CuAlOx/CNTs) and 10% (mass) Cu-Al/c-CNTs (denoted as CuAlOx/c-CNTs)catalysts were prepared by wet impregnation method for comparison,the molar ratio of Cu/Al was fixed at 3:1.In detail,for CuAlOx/c-CNTs catalysts, the treated CNTs power was dispersed in deionized water. Then, citric acid (CA) was dissolved in the above solution by sonication for 5 min. The solution was subsequently added into an aqueous Cu(NO3)2∙6H2O and Al(NO3)3∙9H2O solution under stirring followed by ultrasonic treatment for 25 min. Then,the impregnated samples were dried at 343 K in the oven for 12 h, followed by calcination at 773 K for 5 h in N2atmosphere to obtain the CuAlOx/c-CNTs catalyst,respectively.The preparation method for CuAlOx/CNTs catalyst was similar to that of the CuAlOx/CNTs catalyst except that the mass of CA was 0 mg.
2.2. Catalyst characterization
X-ray powder diffraction (XRD) patterns were carried out on a Rigaku DX-2700 X-ray diffractometer with Cu Kα radiation(λ = 0.15412 nm) to analyze the phase of the LDHs precursor and catalysts. Fourier transform infrared spectroscopy (FT-IR) of the LDHs precursor was analyzed by TENSOR II Fourier transform infrared spectrometer from Bruker, Germany at the range of 4000–400 cm-1.The thermogravimetry(TG)was employed to analyze the thermal stability of LDHs precursor on Synchronous thermal analyzer STA449F3 from Netzsch, Germany. The morphology and surface atomic concentration of CuAl-LDHs/c-CNTs precursor were examined by a scanning electron microscope (SEM, SU8010,Japan) and energy dispersive spectrometer (EDS). Transmission electron microscopy (TEM) analyses were performed on a JEM-2100F (JEOL, Japan) with an accelerating voltage of 200 kV. The N2adsorption/desorption isotherms were measured on the automatic gas adsorption analyzer ASAP-2460 from Micromeritics,America. The specific surface area, pore volume and pore size distribution of the catalysts were calculated using Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halenda (BJH) methods. In detail,samples were degassed under nitrogen atmosphere at 473 K for 4 h, followed by nitrogen adsorption in liquid nitrogen at 77 K.The elemental composition of the catalysts was tested by X-ray photoelectron spectroscopy (XPS) which was carried out on Thermo Fisher ESCALAB 250xi system with Al Kα radiation(hν = 1486.6 eV). To evaluate the redox property and acidity of the catalysts, H2-temperature programmed reduction (H2-TPR)and NH3-temperature programmed desorption (NH3-TPD) experiments were employed on a VDSORB-91i automated chemisorption analyzer, separately. The procedure of H2-TPR experiment was as follows: 30 mg catalysts were pretreated for 1 h at 573 K under a high-purity Ar atmosphere. Then the sample was cooled down to 303 K in the same atmosphere. After that, under 10% (vol)H2/Ar atmosphere (gas flow rate being 10 ml∙min-1), hydrogen consumption was monitored from 303 to 873 K (the heating rate being 10 K∙min-1). The H2-TPR curve was fitted by peak splitting using Origin software, and hydrogen consumption was calculated from the peak area.In case of NH3-TPD,the samples(30 mg)were pre-treated at 423 K for 30 min to remove impurities over the catalyst, cooled to 323 K in a flow of He. Following cooling to room temperature, the sample was treated with NH3(3% (vol) NH3/Ar)for 30 min until adsorption saturation. Finally, NH3-TPD experiments were performed in a He gas stream from room temperature to 873 K(the heating rate being 10 K∙min-1).The data recorded by thermal conductivity detector (TCD) on-line detector was separated and fitted by Origin software, and the NH3desorption and the ratio of strong and weak acid sites were calculated by the normalization method.In situdiffuse reflectance infrared Fourier transform spectroscopy (in situDRIFTS) spectra was performed by a FTIR spectrometer (Bruker Tensor 27). Before each experiment, the catalyst was pretreated in a flow of nitrogen at 573 K for 10 min, after that cooled to the testing temperature. The background spectrum is noted under the N2flow.At last,the spectrum was collected over accumulative 64 scans with a resolution of 4 cm-1.
2.3. Activity measurements
The NH3-SCO activity of the catalysts(0.25–0.42 mm)was evaluated in a fixed-bed reactor (i.d. = 8 mm). The reaction gas was composed of 0.06% (vol) NH3, 5% (vol) O2with N2as the balance gas with a total gas flow rate of 120 ml∙min-1and the gas hourly space velocity(GHSV)was 45000 h-1.The MKS MultiGas 6030 FTIR spectrometer was used to detect the online gas concentrations and the data were recorded as the reaction was in a steady state at each temperature point. NH3conversion and N2selectivity were calculated as follows Eqs. (1) and (2):
The subscripts ‘‘(X)in” and ‘‘(X)out” representing the inlet and outlet gas concentrations of the reactant, respectively.
3. Results and Discussion
3.1. Structural characterization of LDHs precursors.
The XRD patterns and FT-IR spectra of CuAl-LDHs/CNTs and CuAl-LDHs/c-CNTs precursor prepared byin situprecipitation assembly method are shown in Fig. 2(a) and (b). In Fig. 2(a), the reflection peaks located at about 11.8°, 24.3°, 35.4°, 38.8°, 46.6°and 60.1° could be indexed to the (0 0 3), (0 0 6), (0 1 2), (0 1 5),(0 1 8) and (1 1 0) planes of the LDHs (PDF # 37-0630), indicating the successful preparation of the LDHs. The FT-IR spectra of LDHs precursors are shown in Fig.2(b).The samples presented the broad peak around at 3436 cm-1, which could be attributed to the stretching vibration of interlayer water molecules,laminate hydroxyl (—OH) and crystal water. Meanwhile, the peak at 1636 cm-1corresponding to the bending vibration peak of the hydroxyl group in the crystal water inside the LDH [28]. The band around at 1581 cm-1was assigned to the vibrational peak of the carbon skeleton (C=C/C—C) [22]. The peaks occurred at 1391 cm-1and 850 cm-1belonged to the C—O anti-symmetric stretching vibration and bending vibration of, respectively [28]. The peak below 800 cm-1was related to the vibration of M—OH and M—O in the hydrotalcite structure [23]. The analysis of XRD and FT-IR results confirmed the successful synthesis of LDHs, and clarified that the interlayer anion was.In addition,the SEM and energy dispersive spectrometer (EDS) were employed to reveal the real micromorphology and surface atomic concentration of the CuAl-LDHs/c-CNTs in Fig. 2(c) and (d). As shown in Fig. 2(c), the SEM image showed that the CuAl-LDHs/c-CNTs precursor exhibited a uniform flake morphology. In Fig. 2(d), it could be more clearly observed that the molar ratio of Cu/Al was about 2.9 in the CuAl-LDHs/c-CNTs.
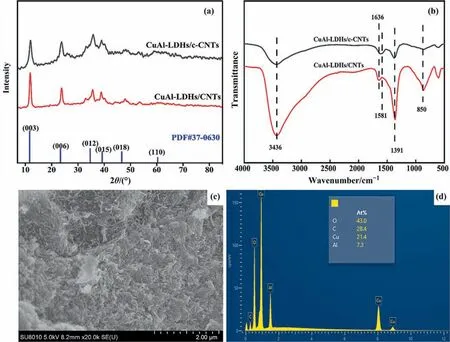
Fig. 2. (a)XRD patterns and (b) FT-IR of CuAl-LDHs/CNTs and CuAl-LDHs/c-CNTs precursors, (c) SEM image and (d) EDS mapping of CuAl-LDHs/c-CNTs precursors.
3.2. NH3-SCO performance for the catalysts.
The NH3-SCO performance of the whole catalysts was tested in the temperature range of 423–603 K with an interval of 303 K as shown in Fig. 3. It was found that the NH3oxidation activity of all catalysts started at about 423 K, and the NH3conversion increased with the reaction temperature rising. Fig. 3(a) and (b)were presented the NH3conversion of CuAlOx/CNTs, CuAlOx/c-CNTs, CuAl-LDO/CNTs, CuAl-LDO/c-CNTs catalysts. In Fig. 3(a),the CuAlOx/CNTs,CuAlOx/c-CNTs catalysts presented similar activity at test temperature range,the NH3conversion was greater than 90% at 543 K. In Fig. 3(b), the CuAl-LDO/c-CNTs catalyst behaved the best activity than CuAl-LDO/CNTs, the NH3conversion of CuAl-LDO/c-CNTs catalyst was above 98% at 513 K. The introduction of CA could be improved the NH3conversion of catalysts.Meanwhile, the NH3conversion of CuAl-LDO/c-CNTs catalyst was higher than CuAlOx/c-CNTs. The N2selectivity of catalysts was shown in Fig.3(c)and(d).It could be observed that the N2selectivity decreased with the increasing of reaction temperature.In Fig.3(c), compared with CuAlOx/CNTs catalyst, the N2selectivity of CuAlOx/c-CNTs catalyst was promoted. For Fig. 3(d), the CuAl-LDO/c-CNTs catalyst exhibited better N2selectivity than CuAl-LDO/CNTs and CuAlOx/c-CNTs catalysts, the N2selectivity was close to 80% in the tested temperature range. The NH3conversion was above 98%and the selectivity was>90%at 513 K for CuAl-LDO/c-CNTs catalyst. It could be seen from Fig. 3 that the catalytic performance of CuAl-LDO/c-CNTs catalyst was better than that of CuAlOx/c-CNTs and CuAl-LDO/CNTs catalysts at the whole temperature range.
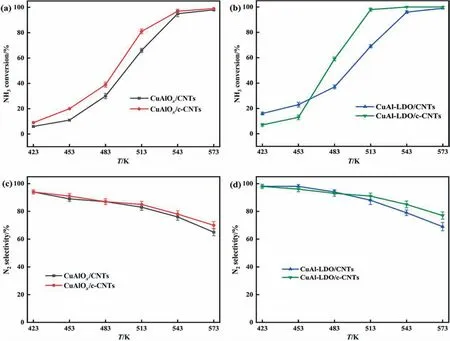
Fig. 3. The catalytic performance of the catalysts: (a) & (b) NH3 conversion, (c) & (d) N2 selectivity.
In general, H2O always presents in vehicle exhaust and industrial flue gas,and the performance of catalyst in NH3-SCO reaction would be suppressed by H2O, especially at low temperatures (below 573 K). In this contribution, 5% H2O was injected to the reaction flue gases. The stability and the resistance to H2O of the CuAl-LDO/c-CNTs catalyst were investigated at 513 K, as depicted in Fig. 4(a). After adding 5% H2O, the NH3conversion of CuAl-LDO/c-CNTs catalyst obviously decreased from 100% to 55% at 513 K after the H2O in flow,because of the competitive adsorption between NH3and H2O. Upon removing H2O from the gas mixture,the conversion was kept at about 80%. It could be suggested that the CuAl-LDO/c-CNTs catalyst possesses satisfactory H2O tolerance performance.In addition,the stability of CuAl-LDO/c-CNTs catalyst was tested by cycle reaction and the result is shown in Fig. 4(b).After five cycles of reaction, the catalyst could still maintain high activity, which could indicate the excellent reaction stability of CuAl-LDO/c-CNTs catalyst.
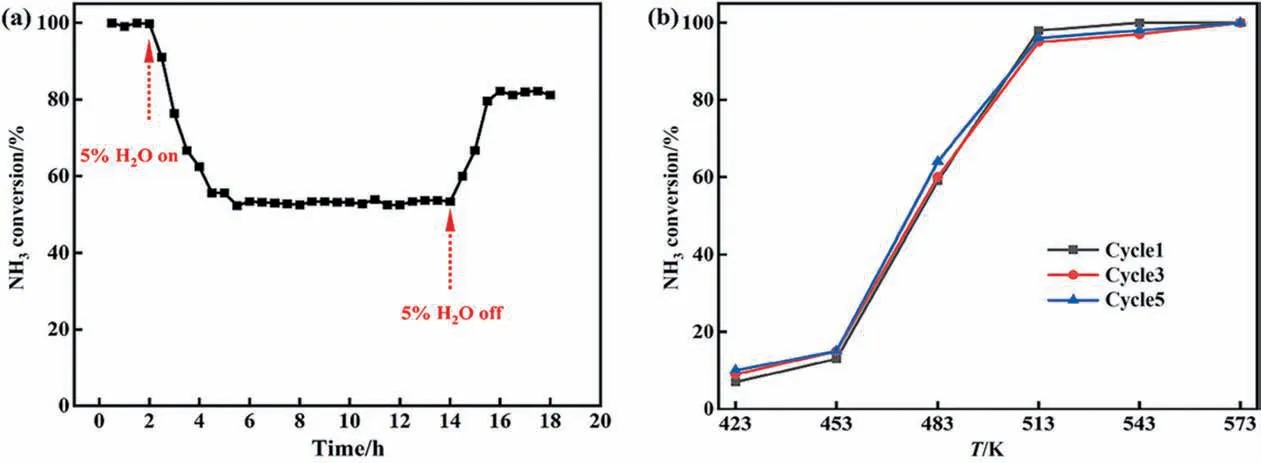
Fig. 4. (a) Effects of H2O on simultaneous oxidation of NH3 over CuAl-LDO/c-CNTs at 513 K. Reaction condition: 0.06% (vol) NH3, 5% (vol) O2, 5% (vol) H2O with N2 as the balance gas, GHSV = 45000 h-1. (b) NH3 conversion as a function of temperature over CuAl-LDO/c-CNTs catalyst in different cycle.
3.3. Structural and property of the catalysts
3.3.1. XRD analysis.
The XRD patterns of the catalysts are displayed in Fig. 5. The CuAlOx/CNTs and CuAlOx/c-CNTs catalysts presented main diffraction peaks at the similar 2θ positions.The peaks at 36.5°and 42.4°corresponded to the(2 1 1)and(2 2 0)planes of the Cu2O(JCPDS#02-1067), respectively. The peaks located at 43.5 and 50.4 were attributed to the (1 1 1) and (2 0 0) planes of the Cu (PDF # 01-1241). The CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts presented four diffraction peaks at 2θ = 35.6°, 38.7°, 48.7°, 61.8°,which were indexed as (0 0 2), (1 1 1), (-2 0 2) and (-1 1 3) of the CuO (PDF # 02-1040). Furthermore, the peaks at 36.5°, 42.4°and 74° corresponded to the (2 1 1), (2 2 0) and (3 3 2) planes of the Cu2O (JCPDS # 02-1067) for CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts,respectively.It was worth mentioning that the peak around at 26.5°for all catalysts in Fig.5 was assigning to the(0 0 2)plane of CNTs[3].In addition,the characteristic diffraction peaks of Al2O3were not detected in the XRD patterns of all catalysts,which might be related to its amorphous state [29].
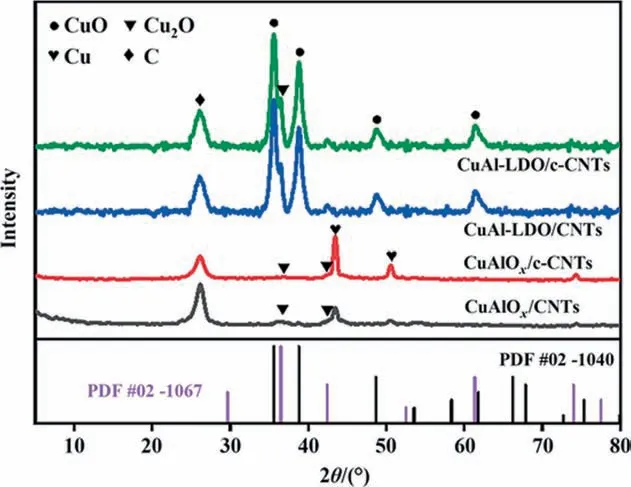
Fig. 5. XRD patterns of the catalysts annealing in N2 atmosphere.
3.3.2. N2 adsorption–desorption analysis.
Textural properties of catalysts were analyzed by N2adsorption–desorption measurements, and the results are presented in Fig. S2. The data of the BET surface area, pore diameter and pore volume were listed in Table 1. In this measurement, the specific surface area and the pore size distribution of the sample are calculated by BET and BJH equations, respectively. In Fig. S2(a), according to International Union of Pure and Applied Chemistry(IUPAC),the CuAlOx/CNTs, CuAlOx/c-CNTs, CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts exhibit a typical character of mesoporous materials. Among them, the N2adsorption–desorption isotherms ofCuAlOx/CNTs and CuAlOx/c-CNTs catalyst were displayed type III and isotherms of CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts were type IV, with H3-type hysteresis at high relative pressure. It could be seen from Fig. S2(b) that the pore size distribution peak of CuAl-LDO/c-CNTs catalyst was the narrowest and sharpest. In Table 1, the BET surface area, pore diameter and pore volume of CuAlOx/c-CNTs catalyst were similar to those of the CuAlOx/CNTs catalyst. The BET surface area of CuAl-LDO/c-CNTs (175 m2∙g-1)catalyst was higher than that of CuAlOx/c-CNTs and CuAl-LDO/CNTs catalysts.The higher surface area of the catalyst is conductive to the exposure of active component and increases its contact possibility with the flue gas, thus leading to enhanced catalytic performance [30]. According to Table 1 and Fig. 3, the CuAl-LDO/c-CNTs catalyst with the best SCO performance has the largest specific surface area. Hence, the surface area of the catalyst has a significant effect on the catalyst activity.
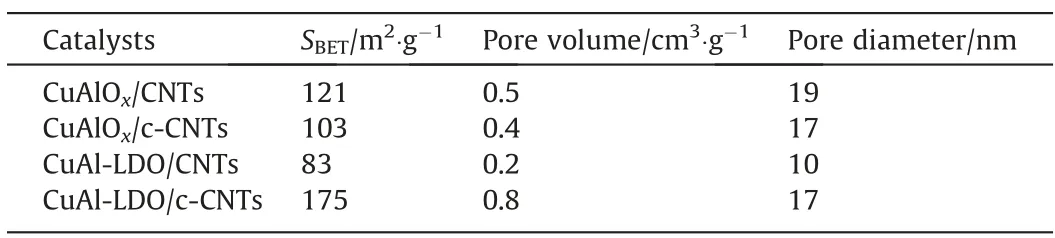
Table 1Data summary from N2 adsorption and desorption of the catalysts
3.3.3. TEM analysis.
The TEM and HRTEM images of (a) CuAlOx/CNTs, (b) CuAlOx/c-CNTs, (c) CuAl-LDO/CNTs and (d) CuAl-LDO/c-CNTs catalysts were shown in Fig. 6. For CuAlOx/CNTs catalyst (Fig. 6(a1)), the metal oxides presented an obviously aggregation state. Compared with other three catalysts,the dispersion state of the active species was improved over the CuAl-LDO/c-CNTs catalyst (Fig. 6(d1)). The CNTs also did not agglomerate over the catalyst.
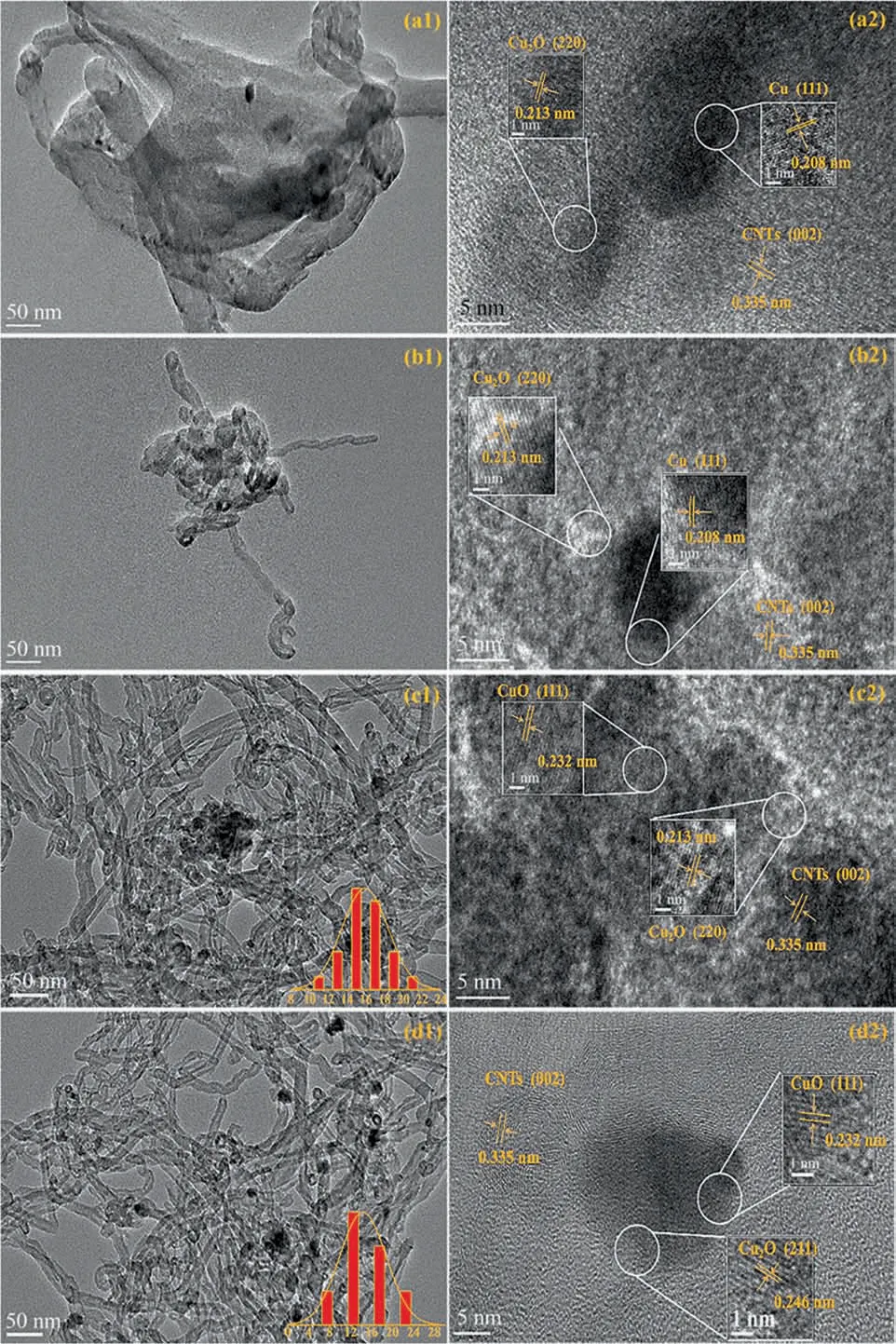
Fig. 6. TEM (a1, b1, c1, d1) and HRTEM (a2, b2, c2, d2) images of the catalysts: (a1, a2) CuAlOx/CNTs, (b1, b2) CuAlOx/c-CNTs, (c1, c2) CuAl-LDO/CNTs, (d1, d2) CuAl-LDO/c-CNTs catalyst.
In the HRTEM patterns (Fig. 6(a2)–(b2)), the lattice spacing of 0.208 and 0.213 nm were observed on the surface of catalysts,which were attributed to the lattice fringe directions of Cu(1 1 1)and Cu2O(2 2 0),respectively.For Fig.6((c2)–(d2)),the lattice spacing of 0.213,0.232 and 0.187 nm could be assigned to the(1 1 1) planes of Cu2O, (1 1 1) and (-2 0 2) plane of CuO, which were identified for CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts.These results were consistent with XRD pattern.The lattice spacing of 0.335 nm could be attributed to the (0 0 2) planes of CNTs over all catalysts. According to the particle size distribution data, it could be seen that the particle size of the active species on the surface of the CuAl-LDO/c-CNTs sample was about 12 nm, which was smaller than the particle size present on the surface of the CuAl-LDO/CNTs (~16 nm). The morphology structure of catalysts has a vital effect on the catalytic performance. In terms of the catalyst activity, the high dispersion of the active species is particularly important.
3.3.4. XPS analysis
The surface oxidation state of copper and oxygen for catalysts was examined using X-ray photoelectron spectroscopy, the corresponding spectra and data summarized in Fig. 7 and Table 2,respectively. In Fig. 7(a), the Cu 2p spectra of all catalysts are detected,and the peaks located at ~932.8 eV and ~953.0 eV,whichwere assigned to Cu 2p3/2and Cu 2p1/2, respectively [31]. Moreover,the Cu 2p3/2peak could be deconvoluted into two peaks corresponding to Cu2+(~935 eV) and Cu+or Cu0(~933 eV),respectively [32]. It was reported that the binding energy of Cu+was similar to that of Cu0. Thus, the XPS spectra of Cu LMM XAES were used to distinguish the Cu+specie and Cu0specie of all catalysts.In Fig. 7(b), the main peak was deconvoluted into three contributions around ~913.0 eV, ~915.6 eV, and ~917.7 eV, which were attributed to Cu+(~913.0 eV), Cu2+(~915.6 eV) and Cu0(~917.7 eV) [31]. For CuAlOx/CNTs, CuAlOx/c-CNTs catalysts, the CuO phase was not observed in XRD pattern (Fig. 5). This might be because the CuO phase exists in the amorphous form on the catalyst surface. For CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalyst,the Cu phase was detected in Cu LMM XAES. The relative content of Cu2+and Cu+species for all catalysts is summarized in Table 2.The ratio of Cu+/(Cu0+ Cu2++ Cu+) was increased as follows:CuAlOx/CNTs (35%), CuAlOx/c-CNTs (36%), CuAl-LDO/CNTs (42%),CuAl-LDO/c-CNTs (45%). When citric acid was introduced into the CuAlOx/CNTs catalyst,the relative contents of Cu+increased.Compared with CuAlOx/c-CNTs catalyast, when Cu and Al were compounded in the form of LDHs and grafted onto CNTs with the help of citric acid, the relative contents of Cu+increased significantly. According to literature, more Cu+was more conducive to the generation of N2.Therefore,it was the main reason for the optimal N2selectivity of the CuAl-LDO/c-CNTs catalyst.

Table 2The XPS results over the different catalysts
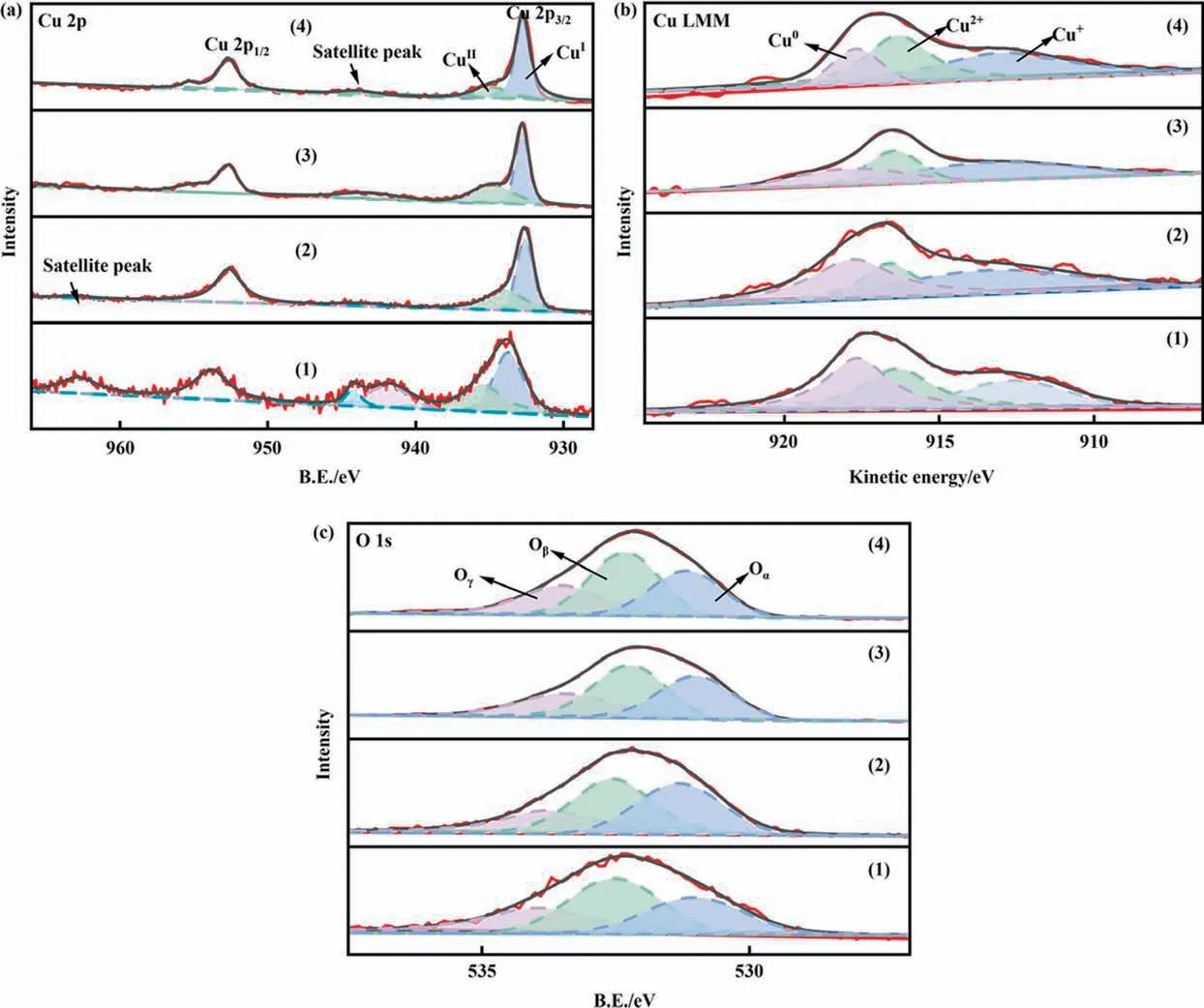
Fig. 7. XPS spectra of the catalysts: (a) Cu 2p, (b) Cu LMM XAES and (c) O 1s. (1) CuAlOx/CNTs, (2) CuAlOx/c-CNTs, (3) CuAl-LDO/CNTs, (4) CuAl-LDO/c-CNTs.
The O 1s spectra for all catalysts are shown in Fig. 7(c), which could be deconvoluted into three sub-peaks.The first peak located at ~531 eV was attributed to the lattice oxygen (O2–, denoted as Oα) [33]. The second peak around at ~532 eV (denoted as Oβ)was assigned to the surface chemisorbed oxygen (O–, O2–and) [34], and the last peak located at ~533.7 eV was allocated to surface oxygen of adsorbed molecular water or hydroxyl species(Oγ) [33,35]. The result of Oβ/(Oα + Oβ+ Oγ) ratio could be calculated by the integral peak area and are presented in Table 2. From Table 2,the Oβ/(Oα+Oβ+Oγ)ratio were similar for different samples.It was reported that the surface chemisorbed oxygen(Oβ)has high mobility and reactivity,which could be favorable for the NH3-SCO reaction[35].On the other hand,the surface chemisorbed oxygen (Oβ) with higher mobility can positively participate in the NO oxidation process to produce NO2and facilitate the‘‘fast SCR”reaction. Therefore, it was not the main reason affecting the catalytic performance of the catalyst.
3.3.5. H2-TPR analysis
The redox property of the catalysts was studied by H2-TPR experiments as displayed in Fig. 8. All catalysts exhibited two reduction peaks at the temperature range of 373–673 K. For CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts, two peaks were observed at 473–673 K related to the reduction of highly dispersed copper oxide and reduction of small crystalline CuOx, separately[23,36]. For CuAlOx/CNTs and CuAlOx/c-CNTs catalyst, there was only one reduction peak at 534 K and 505 K, which could be ascribed to the reduction of highly dispersed copper oxide. Compared with the CuAlOx/CNTs catalyst, the reduction peak of other three catalysts all shifted to lower temperature, which means that the redox capacities of the other three catalysts were improved. It is well known that the lower the peak reduction temperature of the catalyst, the higher the surface oxygen activity [37]. The temperature of the reduction peak was consistent with the NH3-SCO performance of all catalysts in Fig. 3. Thus, the CuAl-LDO/c-CNTs catalyst displayed better redox property, which was favorable for the oxidation of NH3,and thus exhibited excellent catalytic performance. The H2consumption of different catalysts was calculated and the results were showed in Table 3. Compared with the CuAlOx/CNTs and CuAlOx/c-CNTs catalysts, the amount of H2consumption was higher for CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts. The H2consumption of CuAlOx/c-CNTs and CuAl-LDO/c-CNTs catalysts was slightly lower than that of the CuAlOx/CNTs and CuAl-LDO/CNTs catalysts, which was attributed to the formation of more low valence state Cu species due to the carbothermal reduction of citric acid.
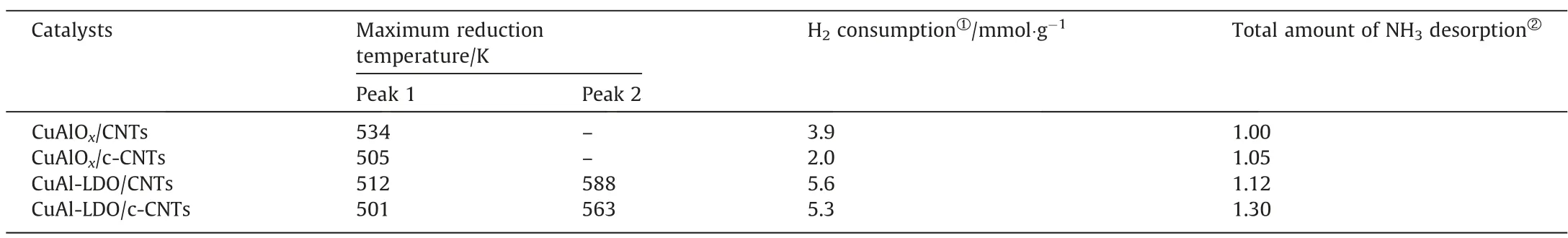
Table 3H2-TPR analysis results and amount of NH3 desorption of catalysts
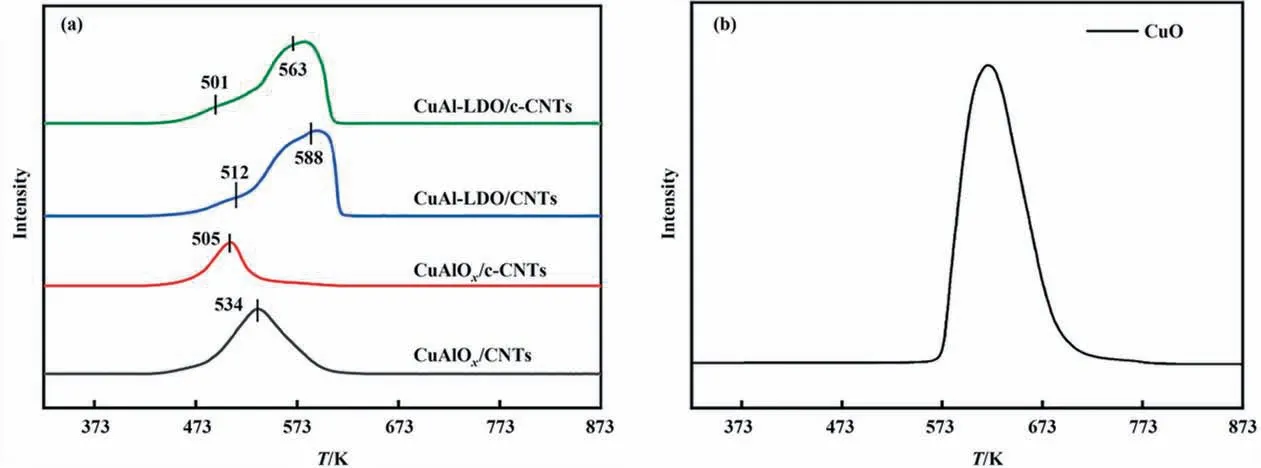
Fig. 8. The H2-TPR profiles of the (a) catalysts and (b) CuO standard sample.
3.3.6. NH3-TPD analysis
As an essential property of NH3-SCO catalysts,the surface acidity of catalysts was evaluated by the NH3-TPD experiments as shown in Fig. 9. For all catalysts, three obvious desorption peaks were observed within 323–873 K in NH3-TPD profiles. The peak located at <473 K could be originated from the adsorbed NH3species desorbed from the weak acid sites. The band around at 473–673 K could be attributed to the desorption of medium strong chemisorbed NH3,and the peak located at>673 K was attributed to be the desorption of strong chemisorbed NH3. The relative total acid amount of catalysts were calculated by the area of its desorption peak, and the corresponding results are normalized and listed in Table 3. Compared with CuAlOx/CNTs catalyst, the addition of CA element could not improve the amount of acid sites of CuAlOx/c-CNTs, but the proportion of acidic sites of different intensities was different. Compared with CuAlOx/c-CNTs and CuAl-LDO/CNTs catalysts, the amount of acid sites of CuAl-LDO/c-CNTs catalysts was improved. While the best NH3-SCO catalyst of CuAl-LDO/c-CNTs displayed the largest number of acid sites.
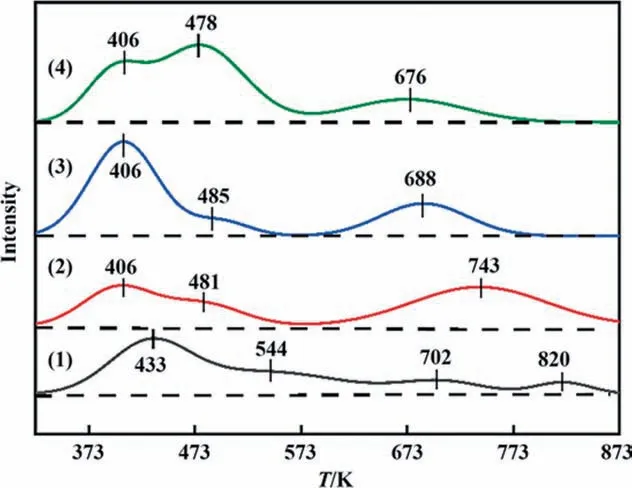
Fig. 9. The NH3-TPD profiles of the catalysts: (1) CuAlOx/CNTs, (2) CuAlOx/c-CNTs,(3) CuAl-LDO/CNTs, (4) CuAl-LDO/c-CNTs.
3.3.7. Kinetic analysis
NH3-SCO kinetic rate measurements over the catalysts were carried out in a quartz fix-bed microreactor.The feed gas composition consisted of 0.06% (vol) NH3, 5% (vol) O2and balance N2. The eigen activity of the catalysts were acquired when the conversion rate is lower than 20%, which eliminates the matter of mass and heat transfer. The kinetic steady-state rate measurements were obtained in the temperature range of 423–513 K. The NH3-SCO reaction rate over CuAlOx/CNTs, CuAlOx/c-CNTs, CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts were calculated by Eqs. (3) and(4) [38].
where the reaction rate constant (ml∙g-1∙s-1) was presentedk, the total gas flow (ml∙s-1) was indicated asV, the mass of catalyst (g)was expressed asW, the NH3conversion (%) was represented asx,Eapresented the apparent activation energy of catalyst (kJ∙mol-1),Rpresented the gas constant(8.314 J∙mol-1∙K-1),Twas the reaction temperature(K)andAwas the pre-exponential factor(mol∙g-1∙s-1).
The Arrhenius plots of NH3-SCO reaction rates over catalysts were exhibited in Fig. 10. The apparent activation energy (Ea) in the NH3-SCO reaction over the four catalysts were decreased in the following order: CuAlOx/CNTs (67.0 kJ∙mol-1) > CuAlOx/c-CNTs (61.0 kJ∙mol-1) > CuAl-LDO/CNTs (49.0 kJ∙mol-1) > CuAl-LD O/c-CNTs (41.7 kJ∙mol-1), which was determined from the slope of the linear plot of ln(k)versus 1000/Taccording to Arrhenius law.
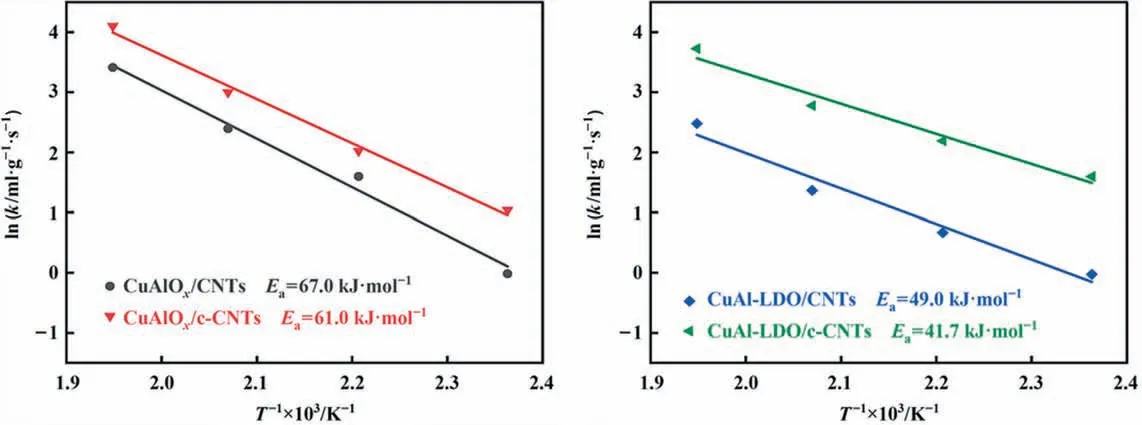
Fig. 10. Arrhenius equation fitting of NH3 kinetic data. Reaction conditions: [NH3] = 0.06% (vol), [O2] = 5% (vol), GHSV = 800000 h-1.
The lower reaction energy barrier is more conducive to the NH3-SCO reaction, thus demonstrating a more excellent NH3removal efficiency.On the whole,due to the improved dispersion of copper species and the increasing exposure of active substances, the apparent activation energy of catalysts decreased significantly.
Reaction conditions:[NH3]=0.06%(vol),[O2]=5%(vol),GHSV=800000 h-1.
3.3.8. In-situ DRIFT analysis
NH3adsorption.Thein situDRIFTS spectra of NH3adsorption at different temperature over CuAl-LDO/c-CNTs catalysts were showed in Fig. 11. For CuAl-LDO/c-CNTs catalyst, several bands at 1745,1691,1645,1544,1514,1456 and 1398 cm-1were observed over catalyst at 423 K. The bands at 1745, 1691, 1645, 1514,1398 cm-1were assigned to symmetric and asymmetric modes of NH4+ions bound to Brønsted acid sites [30,39–42]. The bands at 1544 cm-1ascribed to the wagging vibrations of amide(—NH2) species [37,43]. The band appeared around 1456 cm-1,which could be ascribed to the —NH in amide species [30,43,44].Notably, these peaks first increased and then decreased sharply as the temperature increased from 423 K to 513 K. At 513 K, the NH4+ions species at Brønsted acid sites were detected (1669 and 1398 cm-1) [43]; correspondingly, new peaks located at 1723,1593, 1069 cm-1were detected at 513 K, which attributed to and the NH3coordinated with the Lewis acid sites (1723, 1593 and 1069 cm-1) [30,45]. The peak at 1456 cm-1was shifted to 1480 cm-1, assigned to the —NH species. The intensity of Lewis acid sites was increased from 513 K to 543 K. In Figs. 11 and S3,the intensity of Lewis acid sites over CuAl-LDO/c-CNTs catalyst was stronger than that of CuAl-LDO/CNTs catalyst.It was considered to better NH3adsorption ability of CuAl-LDO/c-CNTs catalyst. Thus,more NH3species coordinated on Lewis acid sites were observed over the CuAl-LDO/c-CNTs catalyst surface than NH4+ions bound to Brønsted acid sites at high temperature. It was concluded the coordination NH3of Lewis acid site was stronger than that of Brønsted acid sites.These results indicated that the NH3(ad)could be activated to form —NH2and —NH intermediates by dehydrogenation at 513 K (reactions (5)–(7)) [40,43,46].
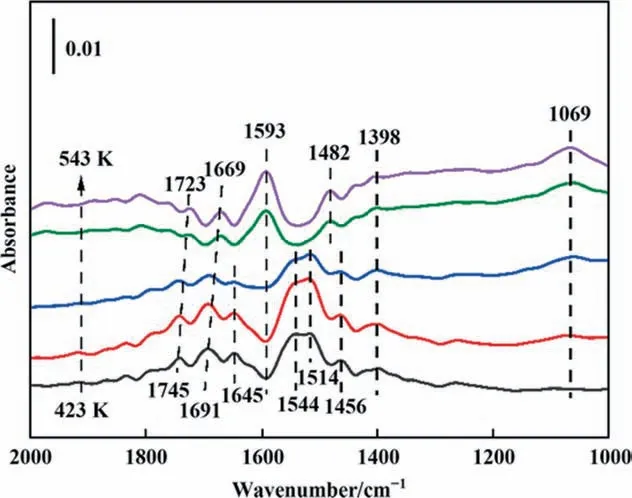
Fig. 11. In-situ DRIFT spectra of NH3 adsorption over CuAl-LDO/c-CNTs catalyst from 423 to 543 K in 30 K temperature intervals.
O2sorption on the catalysts with preadsorbed ammonia.In situDRIFTS spectra of O2adsorption with pre-absorbed NH3of CuAl-LDO/c-CNTs catalyst at 513 K were analyzed in Fig. 12. Firstly,the catalysts were treated with NH3/N2for 30 min at 513 K, followed by purging with N2for 10 min. Then O2/N2was passed into the reaction tank, and the time-varying spectra were collected for 30 min.When CuAl-LDO/c-CNTs catalyst was pre-absorbed NH3for 30 min at 513 K, it was clear that the bands (1745, 1693, 1634,1519, 1464, 1389, 1256 and 1085 cm-1) could be attributed toions bound to Brønsted acid site (1745, 1693, 1634, 1519,1389 cm-1) and asymmetric and symmetric bending vibrations of the N—H bond in NH3coordinated with the Lewis acid sites(1256 and 1085 cm-1)[30,43],respectively.In addition,the bands at 1546 and 1345 cm-1were assigned to the amide(—NH2)species[47]. The band about 1464 cm-1was assigned to the —NH species[44,47,48]. After the introduction of oxygen flow, the intensity of these bands as for NH3at Lewis acid site decreased rapidly and could not be detected until 5 min.And the intensity of bands about—NH2(1345 cm-1) and —NH (1464 cm-1) species were also decreased. However, the band intensity ofat Bronsted acid site did not change with the introduction of oxygen. The intensity of the band at 1519 cm-1was increased with the production of monodentate nitrates, and the intensity of the peak at 1464 cm-1was increased with the production of nitrosyl(—HNO)specie when the introduction of oxygen flow 1 min, and then the intensity was decreased which was meaning the consumption of —HNO specie[43,44]. Meanwhile, the bands around at 1389, 1345 cm-1were shifted to 1407, 1322 cm-1with the extension of oxygen supply time, which were assigned to nitrite species (1407 and 1322 cm-1) [46,49]. In addition, the O2adsorption with preabsorbed NH3over CuAl-LDO/CNTs catalyst investigated by usingin situDRIFTS (Fig. S4). In Fig. 12 and Fig. S4, the redox reactions between NHxand active O atoms formed NOx(reactions (8)–(14))over CuAl-LDO/CNTs and CuAl-LDO/c-CNTs catalysts [46]. The formation of nitrate and nitrosyl was accompanied by the consumption of NH3at the L-acid site, NH2, and NH. It was consistent with the decrease of NHxspecies band intensity and the increase of nitrate band intensity.
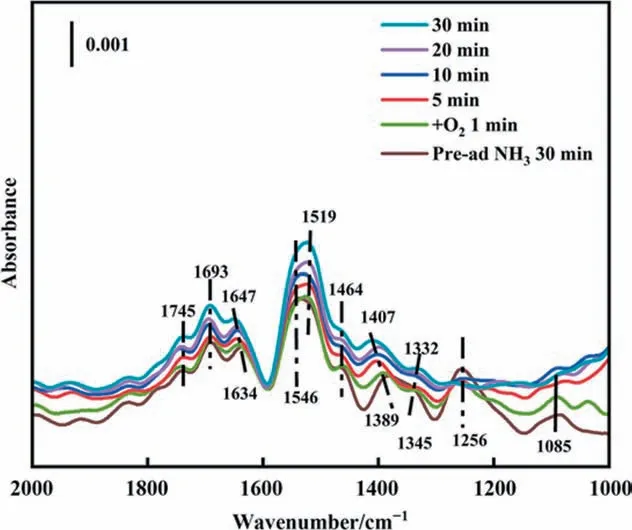
Fig. 12. In-situ DRIFT spectra of O2 adsorption with pre-absorbed NH3 over CuAl-LDO/c-CNTs catalyst at 513 K.
Co-adsorption of NH3and O2.In situDRIFTS spectra of NH3+O2adsorption at 513 K over CuAl-LDO/c-CNTs catalyst are presented in Fig. 13. When NH3+ O2was injected simultaneously over CuAl-LDO/c-CNTs catalyst, peaks at around at 1618, 1377, 1267 and 1140 cm-1were assigned to the NH3coordinated with the Lewis acid sites[45,46].Theions bound to Brønsted acid sites could be observed at 1677 cm-1[41]. In addition, the band appeared around 1459, 1344 and 1086 cm-1could be ascribed to the —NH and —NH2species [47]. The peak around at 1538 cm-1corresponded to the monodentate nitrate species[43]. The intensity of these bands increased with the reaction gas purging time increasing. With the prolonged exposure time to reaction gas,new band assigned to the —HNO was observed at 1480 cm-1[44,47]. The band at 1459 cm-1was shifted to 1438 cm-1. The band at 1086, 1438 and 1538 cm-1became weaker, indicating the —NHxand the monodentate nitrate species were consumed by reaction.According to the DRIFTS results,the reaction path over CuAl-LDO/CNTs (Fig. S6) and CuAl-LDO/c-CNTs catalysts followed the internal SCR (iSCR) mechanism, indicating that the addition of CA did not change the reaction mechanism, but only enhanced the adsorption ability of NH3over the catalyst. The reaction path over CuAl-LDO/c-CNTs catalyst shown in Fig. 14.
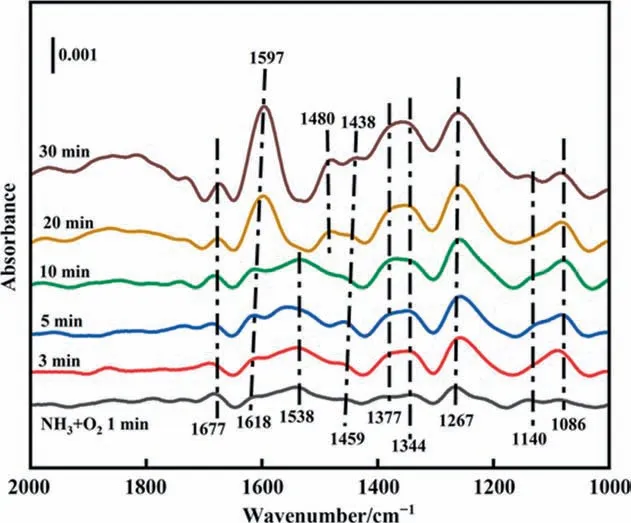
Fig. 13. In-situ DRIFT spectra of NH3 + O2 adsorption over CuAl-LDO/c-CNTs catalysts at 513 K.
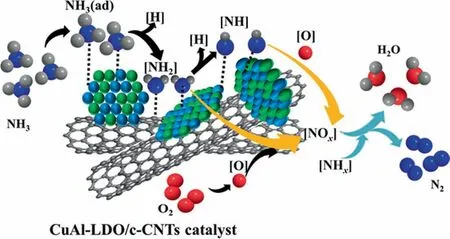
Fig. 14. Reaction pathways of NH3-SCO reaction over CuAl-LDO/c-CNTs catalyst.
4. Conclusions
To sum up,a novel CuAl-LDO/c-CNTs catalyst was fabricatedvia in situoriented assembly of layered-double hydroxides(LDHs)and citric acid-modified carbon nanotubes followed by annealing treatment. The as-prepared CuAl-LDO/c-CNTs catalyst was applied to the NH3-SCO reaction and presented superior catalytic performance (98% NH3conversion with nearly 90% N2selectivity at 513 K). XRD, TEM and XPS analyses manifested that the CuO and Cu2O nanoparticles on the surface of CuAl-LDO/c-CNTs catalyst exhibited more excellent dispersion than CuAlOx/c-CNTs catalyst prepared by traditional wet impregnation method. NH3-TPD and H2-TPR analyses revealed that both the acid sites and redox property of CuAl-LDO/c-CNTs catalyst were improved.The larger specific surface area and better dispersion were conducive to the exposure of more active sites, thus boosting the redox capacity of the active site and NH3adsorption capacity.In situDRIFTS results proved that the iSCR mechanism was performed over CuAl-LDO/c-CNTs catalyst. In short, CuAl-LDO/c-CNTs catalyst presented greater potential than the traditional supported catalyst, and broadened the application of LDO catalyst in the field of lowtemperature NH3-SCO.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51978436, 52000092, 22272116), Fundamental Research Program of Shanxi Province (202103021224043).
Supplementary Material
Supplementary material to this article can be found online at https://doi.org/10.1016/j.cjche.2023.04.019.
 Chinese Journal of Chemical Engineering2023年11期
Chinese Journal of Chemical Engineering2023年11期
- Chinese Journal of Chemical Engineering的其它文章
- Effects of the original state of sodium-based additives on microstructure,surface characteristics and filtration performance of SiC membranes
- Comprehensive analysis on the economy and energy demand of pressure-swing distillation and pervaporation for separating waste liquid containing multiple components
- Esterification of acetic acid with isobutanol catalyzed by ionic liquid n-sulfopropyl-3-methylpyridinium trifluoromethanesulfonate:Experimental and kinetic study
- Numerical investigation of film forming characteristics and mass transfer enhancement in horizontal polycondensation kettle
- COF-derived Co nanoparticles@N-doped carbon electrocatalysts for highperformance Zn-air batteries
- A potential-responsive ion-pump system based on nickel hexacyanoferrate film for selective extraction of cesium ions