金属锰暴露致神经毒性的多组学研究进展
2024-01-09 02:54毕玉洁陈汉清李晓波
中国无机分析化学 2024年1期
毕玉洁 陈汉清 李晓波 陈 瑞
(首都医科大学 公共卫生学院,北京 100069)
锰是人体各种细胞功能中必不可少的微量元素,可作为机体多种酶的辅因子参与酶的功能活动,如谷氨酰胺合成酶以及锰超氧化物歧化酶。膳食是人体摄取必需微量元素锰的主要来源。中国居民膳食指南参考摄入量中提到,成年人锰适宜摄入量(AI)为4.5 mg/d。除自然来源外,采矿、焊接、干电池制造、锰合金生产、锰盐生产、含锰汽油添加剂和农用化学品等人为因素使空气、土壤和水环境中锰浓度显著升高。生物累积进一步导致受污染区域环境和食品中锰的水平增加,从而引发环境暴露人群短期和长期健康风险[1-2]。锰的过度暴露已被证明与人类疾病相关的各种毒性终点有关[3]。环境及职业长期暴露于高锰环境中,将导致锰在大脑基底神经节蓄积,最终引发锰中毒相关神经系统疾病(图1),主要表现为类帕金森综合征的运动功能障碍以及抑郁焦虑等情绪障碍[4-5]。流行病学研究表明,与没有锰排放的城区相比,高锰排放量城区的帕金森病发病率更高[6]。此外,锰暴露量增加对儿童智力和行为功能以及成人认知功能有不利影响,呈负相关,也提示锰暴露还与认知功能障碍性疾病存在关联[7-8]。在职业暴露下,人类大脑中锰浓度可增加1~5倍[9]。进一步的研究表明锰积聚在基底神经节富含多巴胺能神经元的脑区,引起多巴胺能神经退行性疾病[10]。多种啮齿动物、模式生物以及细胞系已被用来探究锰的摄取、稳态和潜在毒性机制[11]。目前研究者已进行了大量的调查和研究,但锰暴露造成的神经精神疾病的分子生物学病因尚未得到详尽阐述,仍需进行更深入的探索。故本文综述近年来金属锰神经毒性的多组学研究,以期为锰的神经毒性机制研究提供理论参鉴。
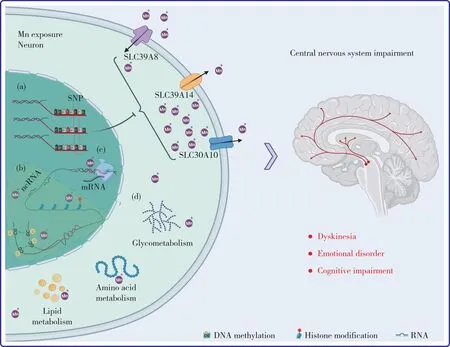
图1 锰暴露诱导中枢神经系统损伤机制示意图a)转运蛋白基因单核苷酸多态性影响锰离子在细胞中的转运作用;b)锰暴露诱导神经细胞DNA甲基化、组蛋白修饰、ncRNA干扰等表观遗传学改变;c)锰暴露诱导神经细胞mRNA转录水平表达差异化;d)锰暴露影响神经细胞葡萄糖、氨基酸、脂质等小分子化合物代谢稳态Figure 1 Schematic diagram of central nervous system impairment induced by manganese exposure.a)Single nucleotide polymorphism of transporter gene affects the transport of manganese ions in cells;b)Manganese exposure induces epigenetic changes such as DNA methylation,histone modification,and ncRNA interference in neurons;c)Manganese exposure induces differential expression of mRNA transcription levels in neurons;d)Manganese exposure affects the metabolic homeostasis of small molecule compounds such as glucose,amino acids,and lipids in neurons.
1 基于基因组学的锰暴露致神经毒性作用机制进展
研究易感因素可以增加对毒物毒性机制的深入了解。遗传突变与疾病发生高度相关且具有绝对风险[12]。遗传性锰转运病的诊断凸显了人体内锰稳态所需溶质载体转运蛋白的重要作用,即参与锰稳态的罕见基因变异可导致细胞内锰稳态失衡。溶质载体家族39成员8(Solute Carrier Family 39 Member 8,SLC39A8)是大脑摄取锰的关键转运蛋白,SLC39A8基因突变导致锰缺乏,其特征是大脑蛋白质糖基化受损[13]。溶质载体家族30成员10(Solute Carrier Family 30 Member 10,SLC30A10)是一种锰外排转运蛋白,其通过将锰离子从胞质转运到细胞外部以防止锰中毒[14]。SLC39A14是一种二价金属离子外排转运蛋白,可以转运锰、锌、铁和镉离子[15]。SLC30A10和SLC39A14协同将锰离子排泄到胆汁和肠道中。即使在没有外部锰暴露的情况下,以上基因的纯合功能丧失突变均影响大脑中锰蓄积,最终导致锰神经毒性相关的进行性肌张力障碍-帕金森综合征[16]。流行病学调查发现,SLC39A8和SLC30A10的常见单核苷酸多态性(Single Nucleotide Polymorphism,SNP)通过锰稳态的差异影响儿童的神经发育(智力、行为)[17]。人类SLC30A10 (rs2275707,rs12064812,rs1776029)以及SLC39A8(rs13107325)的等位基因突变影响不同队列和年龄组健康个体的锰负荷[18]。一项全基因组关联研究显示,SLC39A8(rs13107325,rs13107325)基因连锁不平衡多态性与大脑磁共振成像表型差异相关[19]。动物实验中,SUNUWAR等[20]基于CRISPR/Cas9技术构建了SLC39A8 (Ala-391-Thr)基因敲入小鼠模型,该小鼠模型出现肝脏和肾脏中的锰水平降低,胆汁中的锰水平升高等锰稳态异常表现,进一步提供了体内锰转运蛋白功能改变的证据。此外,阳离子转运ATP酶13A2(ATPase Cation Transporting 13A2,ATP13A2)基因多态性也显著影响锰暴露的意大利老年人运动协调性[21]。
锰转运蛋白基因SLC39A8和SLC30A10已反复验证锰神经毒性的易感性及对机体锰稳态的影响,并且这些基因的常见变体与细胞内锰浓度之间具有环境-基因相互作用关联。然而,关于锰的遗传毒性潜力的研究结果较为局限,需要更广泛的探索性研究来阐明锰的遗传毒性。
2 基于表观遗传学的锰暴露致神经毒性作用机制进展
表观遗传学改变表现为基因表达和调控的遗传变化,即DNA的修饰方式改变,而非DNA序列编码改变,包括DNA甲基化、组蛋白修饰、非编码RNA(Non-coding RNA,ncRNA)干扰,蛋白翻译后修饰等。表观遗传改变可能对机体具有长期影响,对于神经发育和大脑疾病的易感性具有重要意义[22]。
DNA甲基化是在胞嘧啶环的5′碳原子上共价添加一个甲基,该过程大多发生在DNA的CpG岛上,并受到DNA甲基转移酶(DNA methyltransferase,DNMT)家族调控[23]。目前已发现DNA甲基化程度可影响神经干细胞分化发育和神经变性[24]。孕期及产后一段时间的环境是晚年疾病易感性的重要决定因素[25]。BOZACK等[26]分析了孕早期母体红细胞中的锰浓度与脐带血中差异甲基化位点(Differentially Methylated Positions,DMP)和差异甲基化区域(Differential Methylation Region,DMR)的相关性,并检测361名儿童队列中收集的血液锰水平与DMP/DMR在儿童中期(6~10岁)的持续相关性。结果发现锰水平与脐血RNA结合Fox-1同系物1(RNA Binding Fox-1 Homolog 1,RBFOX1)基因的cg02042823位点甲基化增加呈正相关,并且关联显著,但在儿童中期该关联性减弱。在儿童中期,男童和女童血液中仍持续存在与锰相关DMP,但男童和女童的差异DMP中无重复值。以上研究结果提示产前接触锰可能导致婴儿DNA甲基化,部分DNA甲基化可持续到儿童中期,并且这种变化可能具有性别特异性。另一组健康单胎妊娠队列研究了孕期锰暴露和胎盘中DNA甲基化的关联性,结果发现高锰暴露与胎盘核受体亚家族3C组成员1(Nuclear Receptor Subfamily 3 Group C Member 1,NR3C1)基因甲基化增加相关[27]。有证据表明,NR3C1甲基化与神经行为障碍的病理生理学存在表观遗传联系[28]。鉴于趾甲的生长速率和样本长度,其可反映中期时间(月)暴露水平,且趾甲中的金属锰已被证实是不良健康结局的确定风险因素[29]。在一项波士顿地区建立的规范老年研究(Normative Aging Study,NAS)中,研究者观察到87个DMP,15个DMR和1个与趾甲锰显著相关的通路(精子活力),其中有4个匹配基因(MAB21L1、CTNNA3、CNTNAP5、PRMT8)与神经发育和神经退行性疾病相关[30]。动物实验中也存在类似的发现,ICR孕鼠妊娠期(妊娠10 d至产后21 d)通过鼠粮暴露于800 μg/g锰后CpG启动子芯片结果分析发现雄性后代海马齿状回24个基因的启动子区域高甲基化,其中Mid1,Atp1a3和Nr2f1基因的CGI高甲基化和转录下调在成年小鼠中持续存在[31]。锰暴露还增加了p53基因的DNA甲基化,导致小胶质细胞中p53转录减少和促炎因子COX-2下游表达增加,并且这种锰效应可被去甲基化试剂5-Aza-dC消除[32]。
组蛋白乙酰化是最早发现的组蛋白修饰,通过影响核小体的结构稳定性调节基因转录和表达[33]。组蛋白乙酰化失衡与神经退行性疾病有关[34]。锰诱导的神经毒性可通过调节组蛋白乙酰转移酶(Histone acetyltransferase,HAT)和组蛋白脱乙酰酶(Histone deacetylase,HDAC)致组蛋白乙酰化或去乙酰化的表观遗传修饰改变。锰可激活几种HDAC,如星形胶质细胞中的HDAC1-5[35]以及神经元中的HDAC1和Ⅲ类HDAC(Sirtuins,SIRTs)[36]。体外研究表明,锰可以通过动态调节HAT和HDAC活性改变组蛋白乙酰化的稳态,导致人类神经母细胞瘤细胞(SH-SY5Y)和大鼠肾上腺嗜铬细胞瘤细胞(PC12)损伤[37]。体内研究显示,组蛋白去乙酰化酶抑制剂(Histone deacetylase inhibitor,HDACi)通过降低小鼠脑和星形胶质细胞中HDAC活性,使组蛋白乙酰化和基因表达正常化来减轻锰的毒性作用[38-39]。锰暴露降低了在超氧化物歧化酶2(Superoxide Dismutase 2,SOD2)基因启动子区组蛋白H3的27号位点赖氨酸乙酰化水平,造成小鼠认知功能障碍,但抗炎抗氧化药物姜黄素逆转了小鼠上述行为学反应,提示氧化应激可能是锰暴露致组蛋白乙酰化失调的可能机制之一[40]。
此外,表观遗传机制还涉及包括miRNA在内的ncRNA。miRNA是一种小型单链非编码RNA分子,其通过与mRNA杂交调节转录后基因表达,起着翻译抑制或降解目标RNA的作用[41]。锰暴露改变SH-SY5Y细胞中73种miRNA水平,包括靶向ATP13A2,α-突触核蛋白(Synuclein Alpha,SNCA)和成纤维细胞生长因子-20(Fibroblast Growth Factor 20,FGF-20)的miR-4306,miR-7和miR-433,上述miRNA的抑制可减弱锰对SH-SY5Y细胞中靶基因的作用[42-43]。miR-138-5p过表达可通过靶向SIRT1基因抑制锰诱导的SH-SY5Y细胞自噬[44]。此外,锰还可通过lncSh2d3c/mmu-miR-675-5p/Chmp4b/Bax轴诱导神经元凋亡,从而导致锰暴露后小鼠学习和记忆能力受损[45]。
综上所述,锰暴露改变了与神经发生相关细胞群的表观遗传基因调控和编程。DNA甲基化、组蛋白异常修饰以及非编码RNA干扰等在锰引起的神经毒性中均发挥重要作用,但是锰引起上述表观遗传基因调控失调的机制并未在多项研究中得到重复验证,有待进一步论述。
3 基于转录组学的锰暴露致神经毒性作用机制进展
转录组学是研究生物体内所有基因表达水平的一项技术,对于理解基因调控、疾病发生和发展具有重要意义。由于人体神经组织的获取存在较大的限制性,基于神经组织转录组学测序的锰暴露相关神经毒性研究主要采用模式动物及细胞系开展。对锰暴露斑马鱼脑组织单细胞测序结果显示,斑马鱼锰暴露后改变了胆碱能神经元、多巴胺能神经元和γ-氨基丁酸(Gamma-aminobutyric acid,GABA)能神经元细胞类型的比例,进一步伪时间分析表明多巴胺能神经元对锰神经毒性的反应最优先,且KEGG信号通路分析表明,锰暴露主要影响多巴胺能神经元铁死亡通路[46]。另一项斑马鱼锰暴露后单细胞测序的研究结果同样表明多巴胺能神经元损伤,差异表达基因GO富集分析显示,其主要与蛋白质、氨基酸和能量代谢、细胞周期和未折叠蛋白质反应(Unfolded Protein Response,UPR)通路有关[47]。在暴露于锰的秀丽隐杆线虫模型的转录组分析发现氧化应激、抗氧化反应、内质网应激和免疫信号通路参与锰诱导的毒性反应[48]。体外研究发现,暴露于生理浓度锰(10 μmol/L MnCl2)5 h后的SH-SY5Y细胞转录组学研究发现,高尔基驻留蛋白质相关基因(BET1,ADAM10和ARFGAP3)表达增加,影响SH-SY5Y细胞蛋白质分泌途径[49]。而暴露于高剂量锰溶液(100 μmol/L MnCl2)5 h的SH-SY5Y细胞影响氧化磷酸化途径及相关基因(ATP6V1H,NDUFAF5和FABP5)作用破坏细胞能量代谢过程,导致细胞和线粒体能量代谢中断以及氧化应激反应从而加速细胞死亡[49]。
上述研究表明,锰暴露后主要损伤大脑中多巴胺能神经元,但当前研究结论仅限于模式动物及细胞系的研究,并不能反映具有复杂生理过程及机制的大脑。因此,推进基于临床生物学样本或实验动物的转录组学分析进程,进一步阐述锰暴露后人群或实验动物生物样本转录组差异,有利于形成衔接遗传物质与代谢物的微观网络。
4 基于代谢组学的锰暴露致神经毒性作用机制进展
代谢组学是基于对不同机体代谢过程相关的细胞、组织或生物流体(例如血浆或尿液)的内源性低分子量生物分子(通常< 1500 Da)的综合分析[50]。代谢组学作为一种快速和可重复的方法,直接反映与暴露相关的生物事件,在职业健康领域的应用范围逐渐增加。代谢组学位于“组学”级联的底部,与环境的上游输入和基因组的下游输出相关。近年来,代谢组学及其相关技术逐渐应用于锰诱导神经毒性的疾病机制以及药物干预研究,研究者基于不同人群、临床生物学样本以及动物模型开展了多项研究。
ASHRAP等[51]观察到母体血液锰、锌、铅和汞浓度与胎儿不良出生结局(早产以及较短的胎龄)有关。此外,有研究表明母体脂质组成稳态在妊娠期间至关重要,特别是对胎儿发育过程[52]。一项研究发现母体锰暴露与缩醛磷脂酰胆碱(Plasmenyl-phosphatidylcholine,PLPC)和缩醛磷脂酰乙醇胺(Plasmenyl-phosphatidylethanolamine,PLPE)的负相关性最强,特别是含有多不饱和脂肪酸的PLPE[53]。
而针对职业暴露人群的研究,SHEN等[54]利用超高效液相色谱-串联质谱(UPLC-MS/MS)法对接触焊接烟雾工人外周血的代谢物水平进行分析,结果显示焊接工人1)脂质代谢稳态失衡,包括糖皮质激素类(皮质醇、皮质酮和可的松)及酰基肉碱类显著降低,二羟基十八烯酸类(9,10-DiHOME和12,13-DiHOME)显著增加,部分溶血磷脂(10个)、磷脂(8个)和二酰甘油(5个)代谢增加;2)氨基酸代谢增加,包括异亮氨酸,脯氨酸和苯丙氨酸;3)S-(3-羟丙基)巯基甲酸(3-HPMA)增加,上述差异代谢物与炎症状态密切相关。CARTER等[55]运用液相色谱-串联质谱(LC-MS/MS)法对17名接触锰的铸造工人尿液样本进行水性代谢物的靶向测定,发现暴露和未暴露工人之间有7种代谢产物的丰度差异具有显著性(FDR≤0.1),包括n-异丁酰甘氨酸、胆酸、鹅肌肽、β-丙氨酸、甲硫氨酸、N-异戊酰氨基乙酸和苏氨酸,3条氨基酸代谢通路受到严重干扰(β-丙氨酸代谢、组氨酸和甘氨酸代谢、丝氨酸和苏氨酸代谢)。台湾某造船厂的35名男性焊工和16名男性办公室工作人员的尿液1H核磁共振波谱法代谢组学特征分析显示代谢产物,如马尿酸盐、S-硫代半胱氨酸、肌酐、丝氨酸、葡萄糖酸盐、甘氨酸、牛磺酸、氧化三甲胺/甜菜碱、丙酮在焊工中显著增加,而唯一显著减少的代谢物是肌酸[56]。上述代谢物涉及糖代谢、氨基酸代谢、氧化/还原和尿素代谢途径。其中具有抗炎、抑制ROS产生以及抗氧化作用的牛磺酸、甘氨酸、甜菜碱等代谢产物增加,一定程度上反映了焊工体内对环境压力的代偿机制。
环境污染物实验动物暴露模型在一定程度上可模拟人类暴露后的疾病状态,有利于环境污染物暴露后发病机制及防治的深入研究。先前的研究表明,职业锰暴露与多巴胺能功能障碍[57-59]和丘脑GABA水平[60-61]之间存在剂量依赖性关联。ZHANG等[46]对成年慢性(90 d)锰暴露模式动物斑马鱼进行非靶向代谢组学测定,结果显示锰暴露影响参与脂质代谢、氨基酸代谢和糖代谢途径的代谢物水平,这种影响在多巴胺能神经元上更为显著。对SD大鼠单次尾静脉注射氯化锰溶液1.5 mg/(kg·bw)后经电喷雾离子法-离子回旋频率-傅里叶变换质谱法(ESI-ICR/FT-MS)非靶向代谢组学分析显示,大鼠大脑中嘌呤或嘧啶的减少、氨基酸代谢紊乱、还原型辅酶Ⅱ(NADPH)产生减少、谷胱甘肽代谢降低以及四种饱和脂肪酸和六种不饱和脂肪酸的含量降低,可能是由于锰暴露引起活性氧的产生增加,线粒体功能障碍以及脂质过氧化而导致抗氧化防御失衡的总体神经保护作用失活[62]。WANG等[63]采用超高效液相色谱-四极飞行时间质谱联用技术对SD大鼠血浆进行代谢谱分析,识别出与锰暴露有关的36种血浆代谢物,主要涉及嘌呤代谢、氨基酸代谢和脂肪酸代谢。值得一提的是,已有研究证实抑郁症患者的3-甲氧基-4-羟基苯乙二醇硫酸盐(MHPG sulfate)水平降低[64]。与临床实验相一致,MHPG在锰暴露后的大鼠血清中含量减少,提示锰暴露与情绪障碍相关[65]。而另一项研究通过尺寸排阻色谱-电感耦合等离子体质谱法(SEC-ICP-MS)探究了不同暴露形式下血清锰结合形式与脑组织锰浓度的关联性,发现亚慢性锰暴露的SD大鼠及单次尾静脉注射氯化锰溶液的SD大鼠血清锰离子均主要以与高分子量化合物结合的形式存在(约90%),但在亚慢性锰暴露下出现血清锰离子与低分子量化合物(柠檬酸盐锰、氨基酸结合锰)结合增加的转变,并且该形式的锰也更有可能通过血脑屏障转运至中枢神经系统[65]。作者进一步通过ESI-ICR/FT-MS法分析代谢物特征发现仅血清柠檬酸盐锰和无机锰与重要脑代谢物(前列腺素H2、15-羟基二十碳四烯酸、前列腺素B1和氧化型谷胱甘肽)强烈正相关,而非血清总锰浓度,即血清总锰浓度对于锰暴露下的脑部状态的测定在诊断上不具有决定性意义。
各类细胞系是目前研究神经生物学及神经药理学的重要手段,具有来源丰富、干扰因素小、实验条件易控制、评价机制灵活等特点。非靶向代谢组学分析显示,与对照组相比,SH-SY5Y细胞系受锰暴露影响的代谢途径包括谷胱甘肽代谢、嘌呤代谢、甘氨酸、丝氨酸和苏氨酸代谢,其中对谷胱甘肽代谢的影响最为显著,且锰暴露抑制谷胱甘肽代谢途径中谷胱甘肽和谷氨酸的表达[47]。补充谷氨酰胺后,可以有效地提高谷胱甘肽水平,触发UPRmt,并逆转锰暴露引起的氧化还原失衡和线粒体功能的神经毒性损伤[47]。此外,还有研究表明,锰暴露引起SH-SY5Y细胞代谢物腐胺的剂量依赖性增加,腐胺的含量与GABA相关代谢产物呈负相关[66]。高分辨代谢组学(HRM)分析显示,在生理浓度锰(10 μmol/L MnCl2)暴露5 h时,神经保护性氨基酸代谢产物(肌酸、磷酸肌酸、磷酸丝氨酸)增加,而暴露于高剂量锰溶液(100 μmol/L MnCl2)5 h,可使能量和脂肪酸代谢产物(己糖-1,6二磷酸、酰基肉碱)减少,线粒体功能障碍,最终导致细胞死亡[67]。
对于利用代谢组学进行的人群锰暴露研究而言,尚缺乏对日常生活或者职业环境中存在除锰暴露外未知或未测定的其他污染物暴露的评估,如PM2.5或焊接作业环境中存在的其他重金属(铬、镍和铁),如何确定已观察到的差异是由于单一锰暴露造成的,或未测定的其他污染物暴露对观察到的差异产生的影响仍需深入剖析。现有研究表明暴露于含锰污染物主要影响机体的氨基酸代谢、脂质代谢、糖代谢以及嘌呤/嘧啶代谢通路,需要进一步的研究来了解上述通路中差异代谢产物的重要性及其潜在机制,以及特定代谢产物水平的改变是否与锰暴露相关。此外,代谢物也受到各种内部因素的影响,如昼夜节律,生产活动甚至是膳食因素以及其他不断发生的生理过程,如何排除上述因素的干扰得到真实可靠的结果需要更详细缜密的实验设计加以验证。
5 展望
鉴于重金属锰的毒性、生物蓄积性以及长期持久性,环境重金属锰暴露的潜在危害越来越受到人类和动物健康与福祉的关注。锰暴露涉及神经发育的多个相关机制,迄今为止尚不存在治疗锰中毒的单一有效药物,因此仍需进一步研究锰的潜在毒性机制,从而开发使用精准分子靶点来治疗或预防锰神经毒性的药物。此外,由于锰靶向作用于多个基因和蛋白质,多个相关靶点的联合治疗策略可能更有利于锰神经毒性的治疗。然而,仅基于单一组学技术在描述机体锰暴露后生理生化反应的差异化十分有限,阻碍了对锰毒性机制的系统理解。目前的研究得到大量可能的锰暴露生物标志物,种类繁多,但缺乏重复性。故仍需大样本、高层次的临床研究,纳入多种生物学样本(血清、尿液、脑脊液、脑组织等)组学数据进行多层组学整合分析有利于证实和扩展目前的研究结果。此外,机器学习已广泛应用于医药卫生领域,以更完整周密的研究方案与更新的技术方法相互辅助,利用多组学领域、多技术平台的交互验证有效地构建调控网络,可提供对毒性机制的全面理解,这将更加精准地识别锰暴露致中枢神经系统疾病的早期诊断生物标志物,巩固职业病防治防线。
猜你喜欢
小哥白尼(野生动物)(2019年5期)2019-08-27
国际口腔医学杂志(2019年3期)2019-05-31
天然产物研究与开发(2018年2期)2018-04-04
吉林大学学报(医学版)(2015年4期)2015-12-17
医学研究杂志(2015年11期)2015-06-10
发明与创新(2015年33期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
养殖与饲料(2014年10期)2014-02-28
遗传(2014年3期)2014-02-28