
UPLC-MS/MS测定追风透骨丸中马兜铃酸类成分含量△
2024-01-03 09:58王颖刘繁红杨燕丽何筱毅何成军杨蕾袁军戴忠
中国现代中药 2023年10期
王颖,刘繁红,杨燕丽,何筱毅,何成军,杨蕾*,袁军,戴忠
1.四川省药品检验研究院 国家药品监督管理局中成药质量评价重点实验室,四川 成都 610097;2.中国食品药品检定研究院,北京 100050
追风透骨丸是《中华人民共和国药典》(以下简称《中国药典》)2020 年版(一部)收载的品种,是由制川乌、白芷、制草乌、香附(制)等23 味中药粉碎后制成的水蜜丸,用于风寒湿痹、肢节疼痛、肢体麻木[1]。处方中细辛含有马兜铃酸,马兜铃酸是马兜铃科植物中含有的系列结构相似的硝基菲类化合物。由于马兜铃酸暴露造成的中毒事件引发世界范围的广泛关注。其中马兜铃酸Ⅰ被证实有明确的肾毒性、致突变性及致癌性[2]。1999 年英国率先全面禁用含马兜铃酸的中草药及其制品;2001 年美国食品药品监督管理局(FDA)也发布了禁用含马兜铃酸中草药的通告,随后几年欧美及世界许多国家和地区,包括中国台湾和中国香港都先后禁用含马兜铃酸的中草药。2003—2004 年,国家药品监督管理局先后禁止关木通、广防己、青木香的药用;对含有马兜铃、寻骨风、天仙藤和朱砂莲的中药制剂严格按处方药管理;要求细辛只能用根及根茎[3],《中国药典》2020 年版(一部)细辛检查项下规定马兜铃酸Ⅰ限量(不得过0.001%)[1]。此外对含细辛的中成药进行马兜铃酸含量限度研究[4-7]。
查阅文献,液相色谱法(LC)在马兜铃酸类化合物的研究中应用较广,主要用于中药材、饮片及含量较高制剂的测定。《中国药典》2020 年版(一部)细辛和辛芩颗粒均采用LC检测马兜铃酸Ⅰ。此外,由于液质联用仪高灵敏度、高选择性、高通量的优势,也已被广泛应用于马兜铃酸类成分的检测和分析,现行涉及马兜铃酸类化合物的补充检验方法均采用LC-质谱法(MS/MS)[5-11]。追风透骨丸成分复杂,故本研究采用超高效液相色谱(UPLC)-MS/MS对其中马兜铃酸和马兜铃内酰胺2种结构类型的4个成分进行检测研究。
1 材料
1.1 仪器
Triple Quad 5500 型超高效液相色谱串联质谱仪(美国AB SCIEX 公司);Milli-Q 型超纯水仪(美国Millipore公司);CPA225D型电子天平(德国赛多利斯公司)。
1.2 试药
7批追风透骨丸(A企业,编号为A001~A003;B企业,编号为B001~B004)。
对照品马兜铃酸Ⅰ(中国食品药品检定研究院,批号:110746-201912,纯度:99.1%);马兜铃酸Ⅱ(批号:PS010654,纯度:99.98%)、马兜铃酸Ⅲ(批号:PS220701-21,纯度:96.33%)、马兜铃内酰胺Ⅰ(批号:PS010658,纯度:99.69%)均购于成都普思生物科技股份有限公司;乙腈、甲酸、甲酸铵均为色谱纯;甲醇为分析纯;水为超纯水。
川乌、白芷等22 味中药购于荷花池中药材市场,经四川省检验研究院黎跃成主任药师鉴定均为正品。
2 方法与结果
2.1 UPLC-MS/MS分析条件
2.1.1 色谱条件 Waters ACQUITY UPLC HSS T3色谱柱(100 mm×2.1 mm,1.8 μm);以乙腈(A)-0.1%甲酸水溶液(含5 mmol·L-1甲酸铵,B)为流动相梯度洗脱(0~5 min,50%A;5~10 min,50%~70%A;10~12 min,70%A);流速为0.3 mL·min-1;柱温为35 ℃;进样量为1 μL。
2.1.2 质谱条件 采用电喷雾离子源(ESI),正离子模式;扫描模式为多反应监测(MRM);离子源电压:5 500.0V;离子源温度:550.0 ℃;气帘气压力:20 psi(1 psi≈6 894.757 Pa);喷雾器流量:55 psi;辅助加热气:55 psi;各化合物质谱参数见表1。
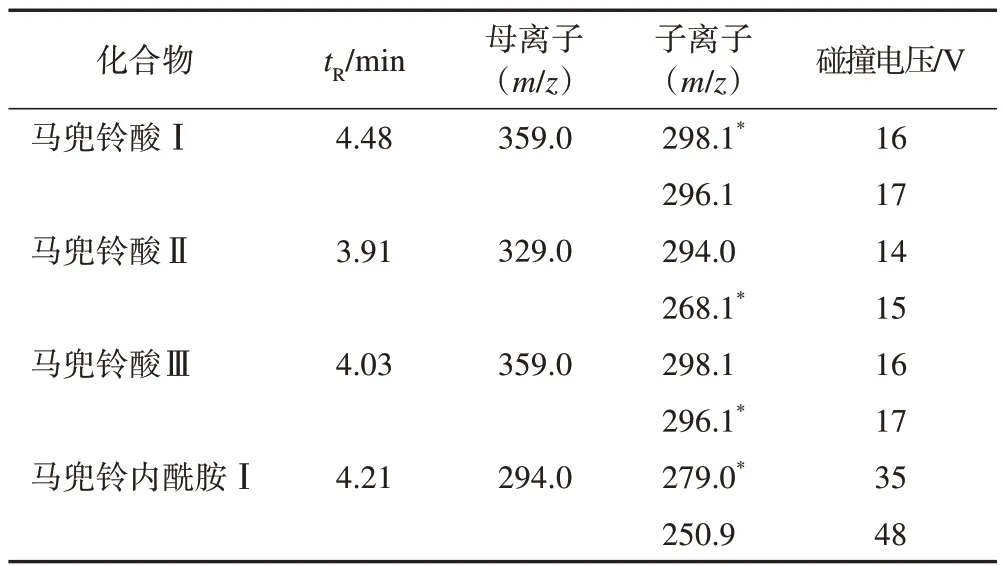
表1 追风透骨丸中待测化合物的质谱检测参数
2.2 溶液的制备
2.2.1 对照品溶液的制备 精密称取马兜铃酸Ⅰ、马兜铃酸Ⅱ、马兜铃酸Ⅲ和马兜铃内酰胺Ⅰ对照品适量,溶于甲醇制成的质量浓度为5 μg·mL-1的混合对照品储备溶液。精密吸取上述对照品储备液,用70%甲醇稀释成质量浓度为10 ng·mL-1的混合对照品溶液。
2.2.2 供试品溶液的制备 取本品适量,研细,取约1.5 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定质量,水浴回流提取1 h,取出,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过(0.22 μm),取续滤液,即得。
2.2.3 阴性样品溶液的制备 分别取细辛以外的22 味中药,按处方比例制备缺细辛阴性样品,按2.2.2项下方法制备缺细辛阴性样品溶液。
2.3 方法学考察
2.3.1 专属性考察 分别取混合对照品溶液、供试品溶液及阴性样品溶液,按2.1项下色谱质谱条件进样,即可得总离子流色谱图(TIC),进行离子对选择后得到各成分的提取离子色谱图(XIC)。结果显示,在马兜铃酸Ⅰ的两对离子通道(m/z359.0→298.1,m/z359.0→296.1)和马兜铃内酰胺Ⅰ的两对离子通道(m/z294.0→279.0,m/z294.0→250.9),供试品溶液中均检出与对照品溶液保留时间一致的色谱峰。在马兜铃酸Ⅱ的两对离子通道(m/z329.0→268.1,m/z329.0→294.0)、马兜铃酸Ⅲ的两对离子通道(m/z359.0→296.1,m/z359.0→298.1),供试品溶液未检出与对照品溶液保留时间一致的色谱峰,阴性样品溶液在各离子通道中均未检出与对照品溶液一致的色谱峰,阴性无干扰,见图1。
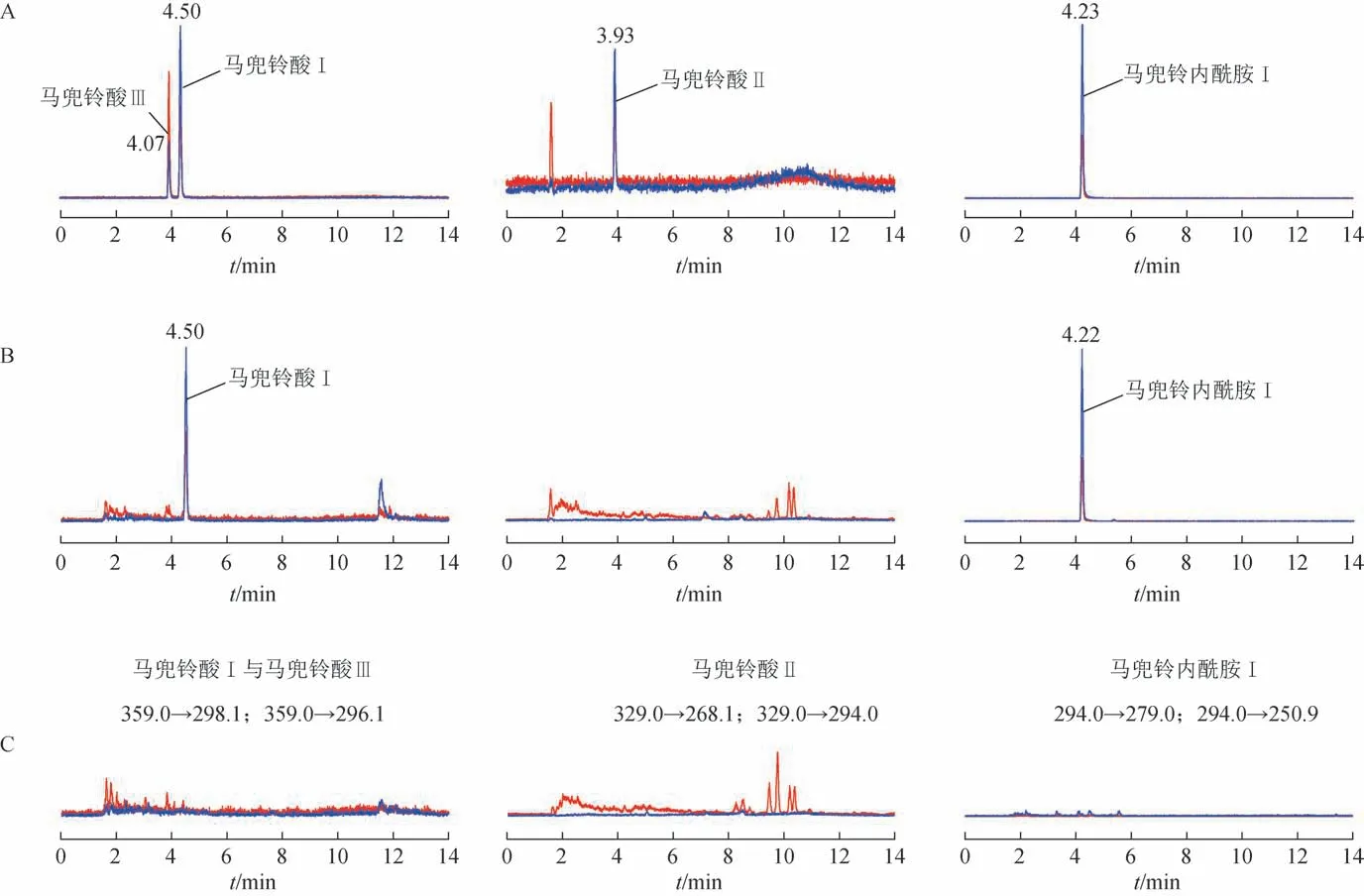
图1 混合对照品溶液、供试品溶液、阴性样品溶液的XIC
2.3.2 线性关系和定量限水平的考察 精密吸取2.2.1项下的混合对照品溶液适量,用70%甲醇稀释成质量浓度分别为2、5、10、25、50、100、250 ng·mL-1的混合对照品溶液。按2.1项下色谱质谱条件进样,以峰面积为纵坐标(Y),质量浓度为横坐标(X)绘制标准曲线,结果4个成分r均大于0.999 5,线性关系良好。计算各离子通道目标峰信噪比(S/N),考虑化合物定性需定性定量两对离子通道均有检出,故确定定量限时定量离子通道按S/N=10计算,定性离子通道按S/N=3计算,以同时满足定量离子通道S/N≥10与定性离子通道S/N≥3的进样质量浓度为定量限。以马兜铃酸Ⅰ为例,质量浓度为2.06 ng·mL-1对照品溶液进样量1 μL时,定量定性离子通道的S/N分别为34.2和12.3,计算定量离子通道S/N=10的质量浓度为0.6 ng·mL-1,定性离子通道S/N=3 的质量浓度为0.5 ng·mL-1,则最低定量水平为0.01 μg·g-1,结果见表2。
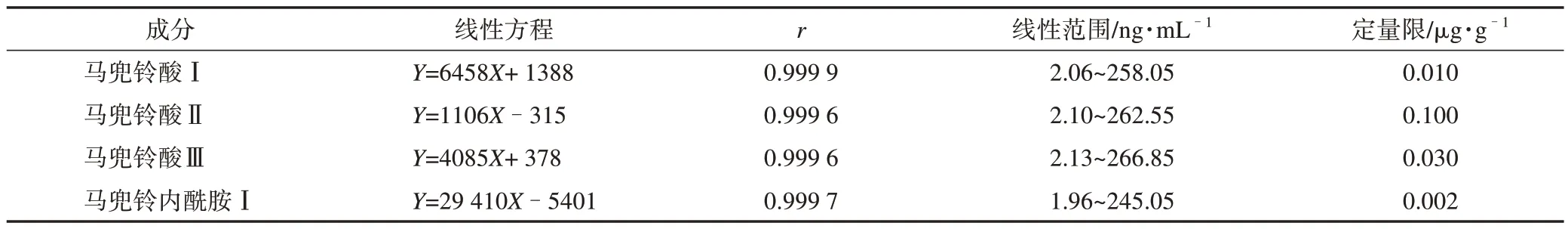
表2 追风透骨丸中4个马兜铃酸成分的回归方程、线性范围及定量限
2.3.3 精密度试验 精密吸取2.2.1 项下混合对照品溶液,用70%甲醇配制10 ng·mL-1的混合对照品溶液,连续进样6次,记录峰面积。结果显示马兜铃酸Ⅰ、马兜铃酸Ⅱ、马兜铃酸Ⅲ和马兜铃内酰胺Ⅰ峰面积的RSD 分别为2.12%、4.32%、1.96%、2.45%,表明仪器精密度良好。
2.3.4 稳定性试验 取同一份供试品溶液,依次于0、3、6、9、11、14、18、21、24 h 进样测定。结果马兜铃酸Ⅰ和马兜铃内酰胺Ⅰ峰面积的RSD 分别为2.44%、2.30%,表明供试品溶液在24 h 内稳定性良好。
2.3.5 重复性试验 取粉碎混匀的样品,平行6份,按2.2.2项下方法制备供试品溶液,按2.1项下色谱质谱条件进样分析。结果未检出马兜铃酸Ⅱ、马兜铃酸Ⅲ,检出马兜铃酸Ⅰ和马兜铃内酰胺Ⅰ的平均质量分数分别为0.093 5、1.066 0 μg·g-1,RSD 分别为2.42%、2.67%,表明该方法重复性良好。
2.3.6 加样回收率试验 精密称取已知含量的样品粉末取粉碎混匀的样品约1.5 g,平行6 份,分别添加混合对照品溶液100 μL,按2.2.2 项下方法制备供试品溶液,按2.1 项下色谱质谱条件进样分析,计算回收率及RSD,见表3。
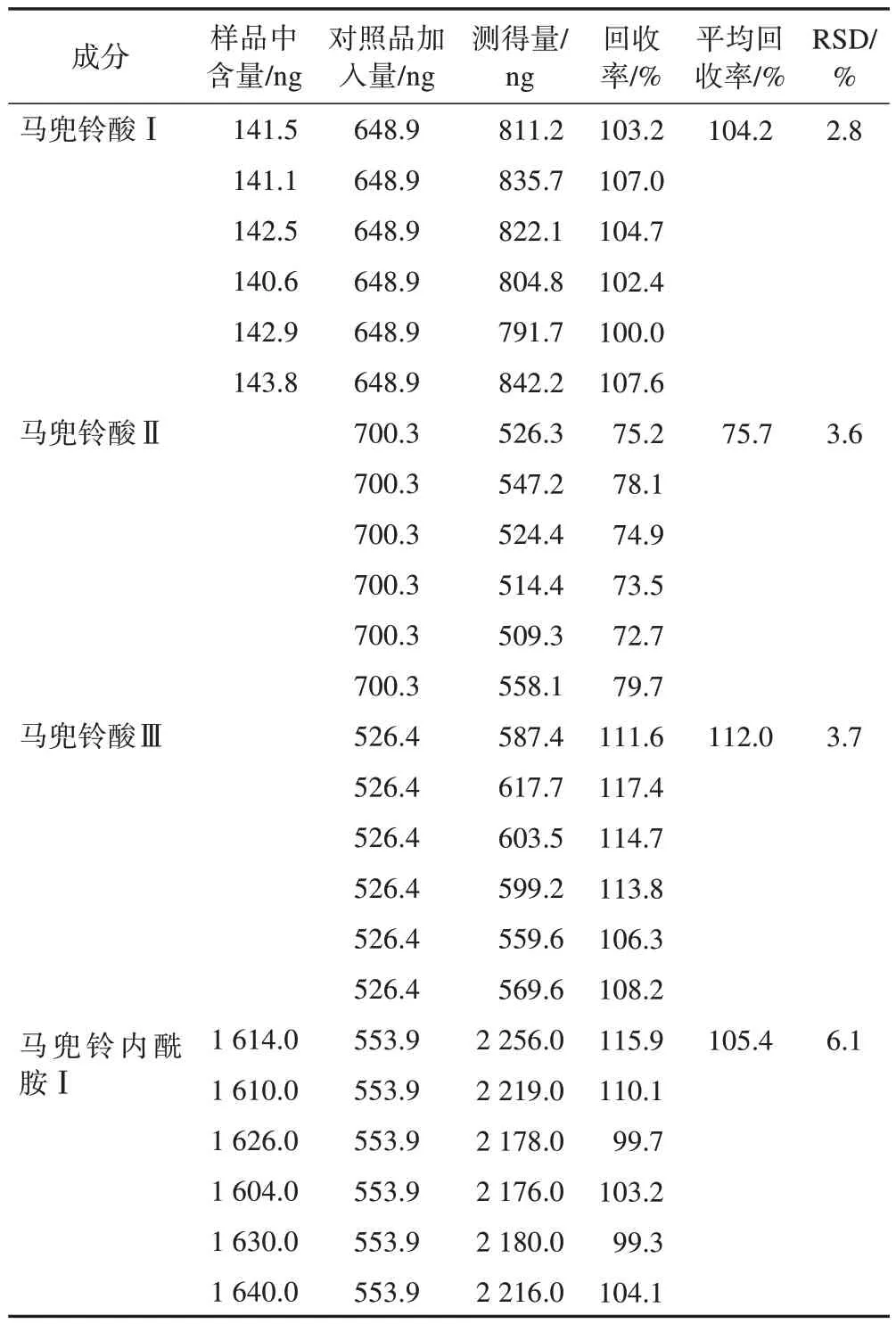
表3 追风透骨丸中4个马兜铃酸成分的加样回收率试验结果
2.4 样品测定
7 批追风透骨丸样品分别按2.2.2 项下方法制备供试品溶液,按2.1 项下色谱质谱条件进样分析,结果马兜铃酸Ⅰ质量分数为0.09~0.41 μg·g-1,马兜铃内酰胺Ⅰ质量分数为0.62~1.30 μg·g-1,样品中均未检出马兜铃酸Ⅱ、马兜铃酸Ⅲ,见表4。

表4 追风透骨丸样品中4个成分质量分数测定结果 μg·g-1
3 讨论
3.1 检测方法的选择
对追风透骨丸处方进行分析,处方中细辛用量为100 g,制成总量为1920 g,加上炼蜜最大制成总量为3072 g,在不考虑转移率的情况下,参考细辛中马兜铃酸Ⅰ的限量,制剂中马兜铃酸Ⅰ理论质量分数不超过0.33 μg·g-1,含量很低;同时由于追风透骨丸处方成分复杂,故选择灵敏度高、抗干扰能力强的基于MRM的LC-MS/MS。
3.2 色谱条件的优化
在进行化合物质谱参数优化时发现,流动相中加入铵盐能提升马兜铃酸类化合物的离子化效率,提高检测灵敏度,使用甲酸铵缓冲液或乙酸铵缓冲液对色谱分离基本没有区别,确定流动相为乙腈-0.1%甲酸水溶液(含5 mmol·L-1甲酸铵)体系。4个成分中马兜铃酸Ⅰ和马兜铃酸Ⅲ互为同分异构体,具有相同的相对分子质量和离子碎片,需要通过色谱柱实现分离后才能检测。分别在Waters ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm)、WatersACQUITY UPLC HSS T3(100 mm×2.1 mm,1.8 μm)、岛津ShimPack GIST C18(100 mm×2.1 mm,2 μm)、Agilent ZORBAX SB C18(100 mm×2.1 mm,1.8 μm)4 种色谱柱上考察对上述2 个成分的分离,结果均获得良好的分离效果。考虑样品采用甲醇直接提取,等度洗脱时,样品溶液可能会存在一些保留较强的极性基质富集于色谱柱中,故洗脱程序采用前段等度、后段梯度的洗脱方式,可最大限度避免和基质干扰物共流出。
3.3 质谱条件的优化
马兜铃酸是硝基菲类化合物,在正离子模式下易得到1 个H+形成准分子离子峰[M+H]+,或加NH4+形成的[M+NH4]+,所以用正离子模式。分别取用4 个对照品溶液直接进样,在ESI+模式全扫描方式下,马兜铃内酰胺Ⅰ得到较高丰度的[M+H]+准分子离子峰m/z294,马兜铃酸Ⅰ、马兜铃酸Ⅱ、马兜铃酸Ⅲ得到较高丰度的[M+NH4]+离子峰m/z359、m/z329、m/z359。在确定母离子的基础上采用子离子扫描方式对子离子的碰撞能进行优化,选择丰度较高的碎片离子为定量离子。
3.4 提取溶剂的选择
结合文献报道[5-7,11],考察不同溶剂对追风透骨丸中马兜铃酸的提取效果。精密称取粉碎均匀的样品适量,分别以甲醇、乙醇、70%甲醇为溶剂,各加入溶剂25 mL 后称质量,超声处理40 min(500 W,40 kHz),放冷后用相应溶剂补足减失的质量,混匀,滤过,取续滤液作为供试品溶液,按2.1 项下条件进样分析。结果显示,对马兜铃酸Ⅰ的提取效率甲醇≈70%甲醇>乙醇,对马兜铃内酰胺Ⅰ的提取效率甲醇>乙醇>70%甲醇。
3.5 方法的准确性
马兜铃酸Ⅰ、马兜铃酸Ⅱ、马兜铃酸Ⅲ和马兜铃内酰胺Ⅰ在20 ng·mL-1添加水平的平均回收率分别为104.2%、75.7%、112.0%、105.4%,RSD 分别为2.8%、3.6%、3.7%、6.1%,表明方法重复性良好,但马兜铃酸Ⅱ的回收率较马兜铃酸Ⅰ、马兜铃酸Ⅲ和马兜铃内酰胺Ⅰ的回收率明显偏低。考虑基质可能造成的影响,比较马兜铃酸Ⅱ定量离子对在纯溶剂中的响应值(A1)与在阴性基质中的响应值(A2),结果显示A2/A1约为1.05,未表现出明显的基质效应,排除基质抑制的影响。研究显示,马兜铃酸Ⅱ在定量限添加水平回收率约为65%,同时在拟定条件下马兜铃酸Ⅱ的质谱响应明显低于其他3 个成分,分析马兜铃酸Ⅱ回收率偏低的原因可能与马兜铃酸Ⅱ的检测灵敏度及较低的加标水平有关[12]。
4 结论
基于本研究建立的方法,实现追风透骨丸中马兜铃酸Ⅰ、马兜铃酸Ⅱ、马兜铃酸Ⅲ和马兜铃内酰胺Ⅰ的检测,结果未检出马兜铃酸Ⅱ和马兜铃酸Ⅲ,检出马兜铃酸Ⅰ和马兜铃内酰胺Ⅰ。马兜铃酸Ⅰ含量基本低于理论值,马兜铃内酰胺Ⅰ含量相对较高。马兜铃酸化合物种类已报道的约178 种,按结构主要分为马兜铃酸类和马兜铃内酰胺类,化合物种类不同毒性差异较大。已有研究表明,马兜铃酸Ⅰ和马兜铃酸Ⅱ具有肾脏毒性和潜在致癌性[13-16],马兜铃内酰胺Ⅰ的毒理学研究尚不充分[17]。有必要首先对具有明确毒性的马兜铃酸Ⅰ建立快速准确的测定方法,研究制定科学合理的限量。
猜你喜欢
中国食品(2020年18期)2020-10-15
山花(2020年7期)2020-07-18
诗潮(2020年6期)2020-06-24
世界科学技术-中医药现代化(2019年7期)2019-10-22
广西中医药大学学报(2019年4期)2019-05-11
癌变·畸变·突变(2016年5期)2016-08-22
中国兽医杂志(2016年5期)2016-06-27
——凹脉马兜铃
广西植物(2016年4期)2016-05-27
中成药(2016年3期)2016-04-06
海南师范大学学报(自然科学版)(2015年2期)2015-12-23
