
Recent development of catalytic strategies for sustainable ammonia production
2023-12-31 04:02SupengYuTingXiangNjudAlharbiBothainaAlaidaroosChanglunChen1
Supeng Yu,Ting Xiang,Njud S.Alharbi,Bothaina A.Al-aidaroos,Changlun Chen1,,
1 Institute of Plasma Physics, HFIPS, Chinese Academy of Sciences, Hefei 230031, China
2 University of Science and Technology of China, Hefei 230026, China
3 Institute of Energy, Hefei Comprehensive National Science Center (Anhui Energy Laboratory), Hefei 230000, China
4 Department of Biological Sciences, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
Keywords:Thermocatalytic ammonia production Electrocatalytic,and photocatalytic ammonia production Thermodynamics process Electrochemistry Multiphase reaction
ABSTRACT Presently,ammonia is an ideal candidate for future clean energy.The Haber-Bosch process has been an essential ammonia production process,and it is one of the most important technological advancements since its invention,sustaining the explosive growth of military munitions industry and fertilizers in the first half of the 20th century.However,the process is facing great challenges: the growing need for ammonia and the demands of environmental protection.High energy consumption and high CO2 emissions greatly limit the application of the Haber-Bosch method,and increasing research efforts are devoted to‘‘green”ammonia synthesis.Thermocatalytic,electrocatalytic,and photocatalytic ammonia production under mild conditions and the derived chemical looping and plasma ammonia production methods,have been widely developed.Electrocatalytic and photocatalytic methods,which use low fossil fuels,are naturally being considered as future directions for the development of ammonia production.Although their catalytic efficiency of ammonia generation is not yet sufficient to satisfy the actual demands,considerable progress has been made in terms of regulating structure and morphology of catalyst and improving preparation efficiency.The chemical looping approach of ammonia production differs from the thermocatalytic,electrocatalytic,and photocatalytic methods,and is the method of reusing raw materials.The plasma treatment approach alters the overall ammonia production approach and builds up a new avenue of development in combination with thermal,photocatalytic,and electrocatalytic methods as well.This review discusses several recent effective catalysts for different ammonia production methods and explores mechanisms as well as efficiency of these catalysts for catalytic N2 fixation of ammonia.
1.Introduction
The Haber-Bosch method,one of the greatest inventions in the 20th century,used for ammonia synthesis derived from a mixture of hydrogen and nitrogen on iron-based catalysts came into being[1–3].It is still the basis for producing more synthetic fertilizers despite the requirement of high reaction temperature and pressure[4].Therefore,as a carbon-intensive industry,the Haber-Bosch process urgently needs to be replaced by a more advanced process that meets the environmental requirements under more moderate conditions[2,3,5].At the same time,ammonia is considered as the most attractive energy carrier due to its light density,carbon-free,easily detectable,high-yield,and high octane rating of 110–130[6].Based on the above situation,more environmentally friendly and mild-condition ammonia production methods with lowtemperature and low-pressure,and even normal temperature and pressure,have been explored.Generally speaking,main methods of ammonia synthesis are: thermal catalysis,electrocatalysis,photocatalysis,and plasma ammonia production.At the same time,chemical looping ammonia production is also derivative from the previous four preparation methods.Thermocatalysis has been continuously developed since the Haber process from the initial optimization of Fe3O4molten iron catalysts to Wüstite (Fe1–xO)catalysts [7].Because of the high activity of Ru metal elements at low temperatures and low pressure,a massive amount of emerging research is divided into many types according to different carriers,promoters,and precursors.Meanwhile,catalyst materials such as bimetallic composites and sporadic transition metals have also been reported [8,9].
Some unavoidable problems with the Haber-Bosch methods,such as high temperatures,high pressures,high pollution,large capital investments,and the uneven distribution of the ammonia industry,have forced scientists to develop simplified ammonia synthesis methods that can be operated under mild conditions.It is no doubt that electrocatalysis is at the forefront of these efforts.Pickett and Talarmin[10]reported the hydrolysis of the nitric compoundcis-[W(N2)z(PMe2Ph)4] to ammonia,which was the beginning of the electrocatalytic ammonia production.This paper summarizes noble metallic catalysts such as Au,Ag,as well as metallic Fe,Ni,Mo,and W-based catalyst and non-metallic Cbased,C-N combined catalysts,B-based,and P-based catalysts.From the point of view of the source,reactant type and morphological structure are crucial factors which affect the difficulty of nitrogen activation and the selectivity,namely the competition between hydrogen evolution reaction (HER) and nitrogen reduction reaction(NRR)[11].Plasma powered by renewable electricity can excite ground state-molecules to active molecule that may react at the surface of selected materials under thermally efficient conditions and at low pressures,which provides an alternative to conventional catalysis and leading to the outlook for the electrification of the chemical industry in future [12].The photocatalytic ammonia production is inspired by nitrogen fixation of photosynthesis.The kinds of photocatalytic ammonia synthesis catalysts are booming in TiO2,Bi base,sulfide,graphite carbon nitride (g-C3N4),layered double hydroxides(LDHs),semiconductor materials,bionic materials and so on.Poor operating stability and inefficient solar energy chemical conversion of photocatalysts are the two most important factors that hinder the efficiency of ammonia synthesis[13,14].The chemical looping ammonia production process,which has been silent for a long time since the invention of Haber-Bosch process,has been revived with the development of new energy sources.Coupling of renewable energy sources such as solar and wind energy with chemical looping processes,simplified and small-scale operations under atmospheric conditions can be achieved.By optimizing the experimental conditions such as reactants,temperature and pressure in each step,the problem of competitive adsorption of N2and H2or H2O can be avoided [15].This review focuses on discussing in detail the evolution as well as opportunities and challenges of various catalytic methods for ammonia production.It is urgently needed for updates on relevant advances since some summaries of ammonia synthesis having fallen far short of the pace of development.The aim of this review is to provide a concise but comprehensive overview of recent advances in the diverse synthesis of ammonia.First,we summarize recent work on the thermochemical ammonia generation and begin with a brief introduction of the mechanisms available for ammonia synthesis.Subsequently,we briefly describe recent advances in the nitrogen fixation capabilities of different types of electrochemical catalysts and even in the reduction of nitropollutants to ammonia.Then we point out the recent advances in photocatalytic catalysts,recent developments in chemical looping and plasma ammonia production are also be pointed out in the final section.
2.The History of Catalyst Development of Ammonia Synthesis
Nicodemus Caro and Adolphe Frank discovered the remarkably simple conversion of cyanide,made by the reaction of calcium carbide and nitrogen (CaC2+N2=CaCN2+C),into ammonia by a water vapor reaction (CaCN2+3H2O=CaCO3+2NH3).The cyanamide process,also named the Frank-Caro process,was the first commercial synthesis of ammonia on a global scale [16].Subsequent to the industrialization of sulphur dioxide oxidation and ammonia oxidation processes for several years,a series of metals active in ammonia synthesis were identified as Os,U,Fe,Mo,Mn,W,etc.In 1910,that German BASF modified Haber method of catalytic ammonia production with Os and U and selected iron catalysts containing Pb and Mg promoters was considered to be one of the first catalytic processes to be applied on a large scale.The new molten iron catalytic system,Fe1–xO system,only broke through the volcanic distribution diagram of activity to iron ratios(with a maximum activity of 0.5),marking a new period of development for ammonia catalysts,with catalysts such as A110-1,A110-2,A201,A301 and ICI3524 emerging [17].Over the last 80 years,although the catalyst formulations have varied and the industrial processes/equipment have been improved and perfected,there has not been much change in the nature of the catalysts,which have not yet been freed from the harsh conditions of high temperature and pressure.
The transition metal electron-donating (EDA) type ammonia catalysts proposed by Tamaru and Ozaki between 1968 and 1971 were not industrialized.By 1970s,the world was becoming increasingly energy-constrained and the cost of ammonia synthesis was rising.In order to reduce costs,countries around the world are committed to the development of low temperature and lowpressure high activity ammonia synthesis catalysts,for example,the British Petroleum and the United States KLG company jointly developed Ru-based ammonia synthesis catalyst[18].It is arguably the second generation of ammonia catalysts after iron catalysts.A suitable active predecessor is selected,a promoter is added and loaded onto the carrier by impregnation,which is transformed into the active component by reduction treatment under certain conditions.There are interactions among Ru,the carrier,the promoter and the facultative ingredients in the catalyst.
Inspired by Fe3O4in the same period,research on nanoscale oxides has flourished and nano-Fe3O4,Fe2O3,CuO,NiO,ZnO,MoO3have been studied successively.Due to the small size and special surface structure of nanomaterials,nano catalysts have special properties.Compared with conventional catalysts,the average selectivity of nano catalysts is 5–10 times higher and the activity is 2–7 times higher.
Since then,transition metal nitrides have been extensively studied as a class of intermetallic compounds with the properties of covalent compounds,ionic crystals and transition metals [19].High-surface-area transition metal nitrides are produced by the programmed temperature nitridation of oxide precursorsviaa‘‘local regularization reaction”under strict nitridation conditions that do not essentially destroy the crystal structure of the oxide precursors.Its surface properties and catalytic performance are similar to those of noble metals such as Pt and Rh,and it is known as a’quasi-Platinum catalyst.In order to break through the thermodynamic limitations,the photoelectric enzyme route to ammonia was explored in the late 1970s,but to date,the different methods still have more or less the same disadvantages of low conversion,high energy consumption and unclear mechanisms.
3.Thermal Catalytic Catalyst for Ammonia Synthesis
Ammonia synthesis,one of the most momentous developments in heterogeneous catalysis field,plays a vital role in the establishment of mechanism and application in many chemical reaction processes.There are two mechanisms can be used to explain how nitrogen molecules be reduced toward ammonia molecules over heterogeneous catalysts: dissociation and association(Fig.1).In dissociation mechanism,the N≡N triple bond is evidently break up leading to the formation of N atoms,which line up at the surface of the catalysts and then combine with hydrogens to form ammonia molecules.Association mechanism can be classified in two categories: associative alternating pathway and associative distal pathway,according to the order of hydrogenation.The typical characteristic of association mechanism is that the nitrogen atoms always are connected until the ammonia has formed,the peculiarity of the first category is that alternating hydrogenation occurs on two nitrogen atoms.When the final single bond has been broken,nitrogen at the far end form ammonia followed by NH2hydrogenation,which finally is liberated.The second type of association mechanism is characterized by the formation of ammonia taking place on the nitrogen atoms away from the catalyst surface by hydrogenation in turn and releasing a metal nitride,subsequent hydrogenation step occurs on another nitrogen atom,forming a second ammonia molecule [2,20,21].Since the N≡N triple bond is strong and is difficult to break,the Haber method,which follows the dissociation mechanism,requires extremely harsh conditions for the reaction to occur[20].For thermocatalytic ammonia production it is overall a variation of the Haber process,only revealing major differences in the catalysts.Multiple thermocatalytic effects have been explored,from ironbased to ruthenium-based catalysts to metal nitride catalysts,as shown in Table 1.
3.1.Molten iron catalyst
Iron catalysts for ammonia synthesis have become one of the most successful and thoroughly studied catalysts in the world[17].The synthesis of ammonia proceeds through several fundamental stages: The process entails the combination of N2and H2,each matched with iron followed by bond cleavage to atomic nitrogen and hydrogen,and then to progressively form ammonia through the transfer of successive hydrogen atoms to nitrogen.Among these basic steps,high bond energy of N2and its dissociative adsorption are the most vital steps[43].The evolution of molten iron catalysts has passed through two main stages.Iron-based catalysts have matured over more than a hundred years.From the initial magnetite base to the later wüstite-based catalysts(both all belong to molten iron catalysts),from iron alloy base to loaded/embedded iron particle carrier composites,the trend in ironbased catalysts has been to move more and more towards reducing the iron content and increasing the interaction of iron with other components,as shown as Fig.2.Conventional ammonia synthesis catalysts were developed in the early 20th century and were prepared from magnetite,which was also the original iron catalysts.Structural promoters were added to the catalytic system with small amounts of non-inducible oxides (usually Al,K and Ca),where Al and CaO were structural promoters and KOxwas an electron promoter to augment the total specific surface area of the catalyst and/or to stabilize the thermal stability of the porous system[44].Traditional iron catalysts with ferric oxide as precursors have been studied extensively for more than 100 years [7].As early as 1926,Almquist and Crittenden [45] investigated the activity of pure iron catalysts (without promoter),then other researchers found that the catalyst at the ratio of Fe2+to Fe3+up to 0.5 had the highest activity [7],with it at the ratio of Fe2+to Fe3+ranged from 0.4 to 0.8 showing the best catalytic activity(Fig.3(a)),which once seemed to be an unquestionable classical conclusion.Apart from that,ammonia synthesis over iron catalysts has a strong structural sensitivity.The order of catalytic activity of iron planes is [111] >[211] >[100] >[210] >[110] [50].In 1986,Liuet al.[17] discovered a new Wüstite-based catalyst based on pyrite(with Fe1–xO as precursor),which was the second major innovation in industrial iron-based ammonia synthesis catalysts.Wüstitebased catalyst displays a typical NaCl structure without magnetic properties and remains stable below 1000 °C,it is famous by the advantage of high activity and low reduction temperature,and complete reaction.In contrast,conventional magnetite-based catalysts possess a sub-stable spinel morphology[7].As shown in Fig.3(b),Liu and Han[7]demonstrated that the trend mentioned above was met at small Fe2+/Fe3+ratios,however,the activity increased again in response to the rise in Fe2+/Fe3+,with the gradual formation of Fe1–xO in the process,which extended the typical volcanic diagram above and overturned the classic conclusion that magnetite was the most reactive.The differences in the precursorand promoter structures lead to changes in the α-Fe active site and crystal plane growth and the formation of internal lattice defects.The sub-stable nanostructures formed by these variations are responsible for their high catalytic activity for ammonia synthesis.Si,Al,K,Ca,and other oxides are often used as promoters in fused magnetite-based catalysts.It is found that the promoter distribution is heterogeneous,where the promoter concentration between the grain boundaries of magnetite was higher than inside the crystal [51].These are where both soluble promoter and glass phases are present,obstructing the tungsten ferrite and calcium ferrite phases.The calcium ferrite between the magnetite particles exhibits an acicular or dendritic crystalline state,which is intertwined with tungsten ferrite without separation.The simultaneous presence of the glass phase occupies the other free space between the magnetite particles [52].Al2O3,K2O,CaO,MgO are the most prevalent promoters in magnetite and tungsten ferrite.In Fe3O4-based catalysts,Al forms substitutional solid solution with Fe3O4,and also forms solid solution FeAl2O4or MgFeAlO4with a small amount of FeO.The uniform distribution of Al2O3is explained by the fact that the solid solution before can form a new solid solution(FeO-Al2O3)-(Fe3O4)again.In Fe1–xO-based catalysts,FeO and Al2O3form a substituted solid solution(FeAl2O4),since the different crystal structures do not further form solid solutions causing Al2O3in the latter less uniformly than in the former [7].K,Al and Ca as the main promoters affect positively the performance and reduction behavior of wüstite-based ammonia synthesis catalysts,narrowing the reduction and disproportionation of sub-stable pyrite,thus allowing a more direct reduction with less magnetite at the final activation temperature [53].Fe1–xO catalysts are still produced by melting method,which has high energy consumption.The wüstite-based catalysts are prepared from natural magnetite with iron as reducing agent and can be melted directly in resistance or electric arc furnaces with controlled temperature to avoid disproportionation reactions(Fig.3(c))[46].The ammonia synthesis process with fused iron catalysts is well established so that Fe3O4/Fe1–xO-based iron catalysts perform well as solids with no extraordinary improvements other than the limited replacement of promoter and adjustments in structure.

Table1 Some generalizations about thermal catalysts for ammonia production

Fig.2. Evolution of iron-based catalysts.

Fig.3. (a) Classical volcano shaped activity curve.(b) Relationship between activities and Fe2+/Fe3+ of catalysts.Reproduced from Ref.[7] with permission of Elsevier,copyright 2017.(c)Preparation of Fe1–xO catalyst by melting method.Reproduced from Ref.[46]with permission of Elsevier,copyright 2020.(d)Construction of Fe3 clusters hosted on the surface of θ-Al2O3(010).Reproduced from Ref.[47] with permission of Nature Communications,copyright 2018.(e) Abbreviated reaction diagram of iron catalysts doped with silicon.Reproduced from Ref.[48]with permission of American Chemical Society,copyright 2020.(f)TiO2-xHy/Fe catalysts overcoming size relationship.Reproduced from Ref.[49] with permission of American Chemical Society,copyright 2020.
In recent years,Nb has been chosen as a potential promoter for ammonia synthesis of tungsten-based catalysts.Nb2O5has lower reduction temperature and faster reduction rate,less inhibition of strong hydrogen adsorption,lower desorption temperature and partially inhibits the formation or separation of solid solutions in the reaction unfavorable to bias or solid solution formation on the catalyst surface.However,the doping of Nb2O5facilitated the sintering of the active phase,while not promoted the growth of the relevant planes [22].The significant growth of single cluster catalysts indicated that there were homogeneous catalytic active centers on non-homogeneous solid surfaces,and authors presented a structure with an active central Fe3 cluster anchored on the surface of θ-Al2O3(0 1 0) (Fig.2(d)) that might be prepared by methods such as the soft-landing cluster method,owing to the fact that triangular Fe3 and conical Fe4 were the most stable clusters on alumina substrates and it was believed that the Fe3 cluster was more stable against aggregation kinetics [47].N2was activated effectively by Fe3/θ-Al2O3(0 1 0),attributing to its multi-step redox capability,large spin polarization and low oxidation state metal.The spin polarized charge was transferred from the 3d orbital of Fe to the π* orbital of N2,and the partially occupied N2β-spin π* orbital reduced the nitrogen bond order conferring the *N2radical property,which leads to N2activation following an association mechanism.The Si-doped Fe-bcc nanoparticles,presenting a Woolf structure,could considerably enhance the reaction by lowering the spin difference between 2 N and 4 N states (Fig.3(e)).The dopant might change the ratedetermining step.Fe-Si binary catalysts were suitable at 2.06 MPa/500 °C or 6.18 MPa/400 °C,substantially better than the harsh industrial conditions(20.6 MPa/500°C),which indicated that the same turnover frequency TOF as the current Haber-Bosch process could be maintained at lower pressure and temperature conditions[48,54].Valence bond analysis revealed that the doping of Si significantly decreases the spin difference between the 2 N and 4 N states compared to pure Fe,stabilizing the 2 N state and leading to a lower potential barrier.Metal-organic frameworks(MOFs)as a high-porosity inorganic–organic hybrid material,have great potential as versatile precursors.Yanet al.[23]proposed that K-promoted Fe/C catalysts with very high Fe loading(>50%(mass))could be prepared by pyrolysis of iron-based MOFs dry gels.K1-Fe-MDC(MOF-derived catalyst)catalysts were composed of spherical metal nanoparticles(NPs)uniformly embedded in a porous carbon matrix.The mass-specific reaction rate could reach 30.4 mmol∙g-1-∙h-1at 400 °C.MOFs were served as a metal and carbon source as well as a sacrificial structure-directed template.The alternating metal sites and organic linkers at the molecular level ensured a favorable dispersion of the metal.The introduction of potassium ions during the preparation process could promote charge transfer from K ions to iron and carbon on the surface,balancing the dissociative chemisorption between H2and N2and suppressing side reactions,thus improving the activity and stability.In bifunctional TiO2–xHy/Fe catalysts,N2and H2were readily dissociated on the iron nanoparticles.The overflow of hydrogen-laden oxygen vacancy (OV-H) on TiO2–xHydestabilized the ‘‘scaling relationship”(Fig.3(f)) between N2dissociation (Ea(N≡N)) and destabilization of NHzintermediates(z=0 to 2),which could vigorously activates the N2catalyst and at the same time hinders the conversion of NHzintermediates [49].
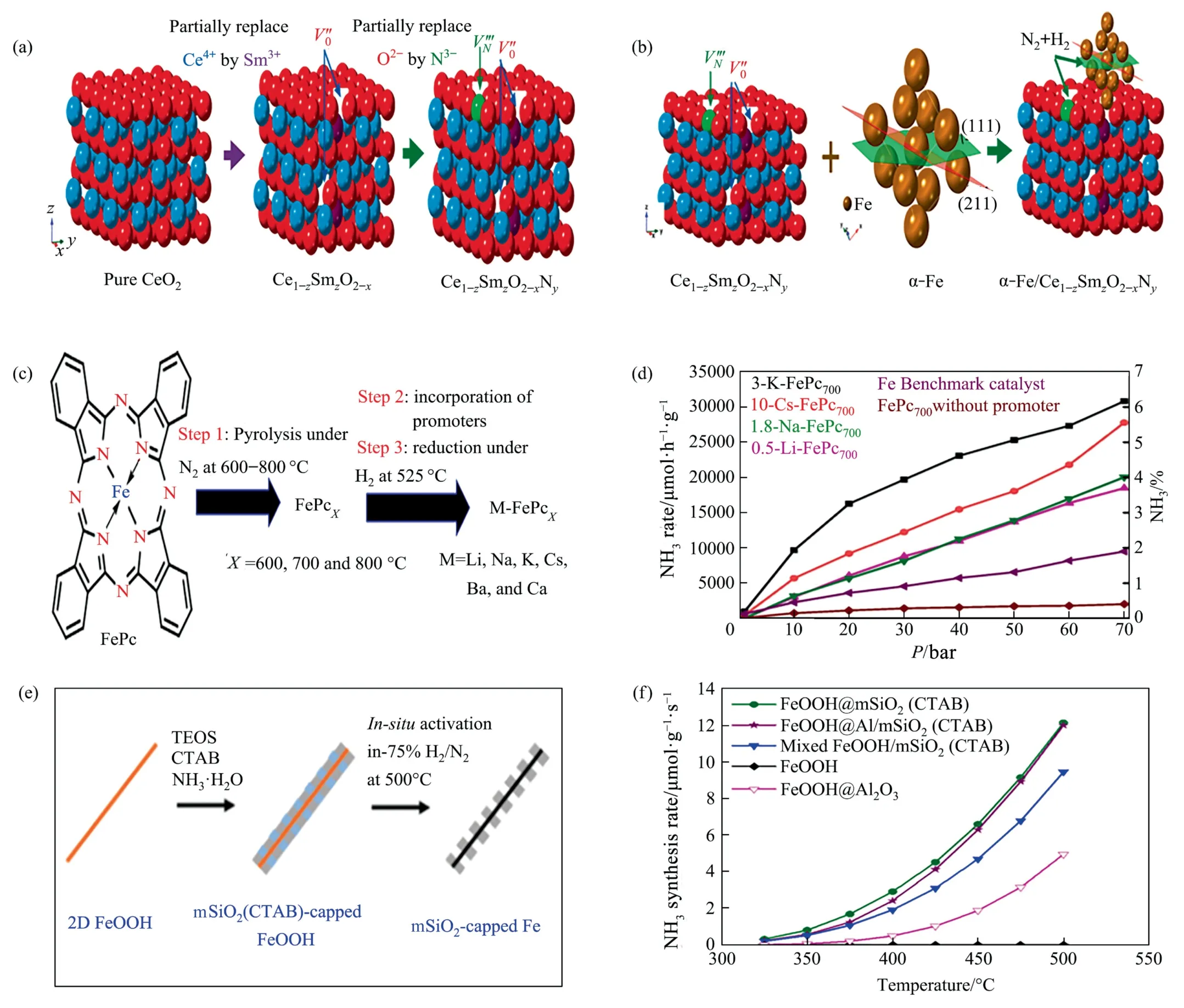
Fig.4. (a)Stepwise formation process of Ce1–zSmzO2–xNy.(b)Shape of Ce1–zSmzO2–xNy loaded with Fe.Reproduced from Ref.[24]with permission of Elsevier,copyright 2021.(c) M-FePcX formation process.(d) Comparison of ammonia production efficiency of FePcX containing different doses of different types of accelerators (1 bar=0.1 MPa).Reproduced from Ref.[25]with permission of Elsevier,copyright 2021.(e)Schematic representation of the fabrication process for the 2D core–shell Fe@mSiO2 catalyst.(f)Fe mass normalized NH3 production rates.Reproduced from Ref.[26] with permission of American Chemical Society,copyright 2021.
Doped oxynitrides with a large number of anionic vacancies,especially nitrogen vacancies,are excellent promoters/cocatalysts for ammonia synthesis.A material with fluorite structure,Ce1–zSmzO2–xNy(z≤0.5),was synthesized by an air combustion method [24].As shown in Fig.4(a),(b),the nitrogen content and the number of anion vacancies increased with higher Sm doping levels and the high concentration of anion vacancies in Ce1–zSmz-O2–xNyfacilitated the nesting/anchoring of Fe particles,resulting in the dependence of strong metal support interaction (SMSI) on stability.z≥0.3 for 80%(mass)Fe-20%(mass)Ce1–zSmzO2–xNycatalysts had an apparent activation energy of about 45 kJ∙mol-1.Based on the phenomenon that the cerium oxide lattice forms more Ce—H species when exposed to H2at high temperatures,the author speculated that the low activation energy and the presence of Sm ions might make the reaction indirectly form H ions through the intermediate of Ce1–zSmzO2–xNy,which might have the same or similar reaction mechanism as TM/BaCeO3–xNyHz.The optimal composition was 80% (mass) Fe-20% (mass) Ce0.5-Sm0.5O2–xNyshowing activity of 18.8 mmol∙g-1∙h-1at 400 °C and 3 MPa.Even when a significant proportion of impurities such as oxygen were injected in the reaction gas,the activity still reaches 70%.Phthalocyanine-based catalysts for ammonia synthesis have also been investigated.Maksoudet al.[25] produced iron-based catalysts by FePc pyrolysis shown in Fig.4(c),promoters were introduced and a constant molar content of alkali metal was ensured to be 0.77 mmol∙g-1.Spherical iron nanoparticles were evenly loaded on nitrogen-doped carbon,and the particle size was temperature-dependent.Carbon and alkali metals forming a multicoated structure in the shell were indistinguishable.Fe was located in the core,while Cs,C,and O were distributed on the shell.This close contact between Cs and Fe was likely to trigger strong metal promoter interactions,which might be the source of enhanced catalytic activity.They also partially suppressed the methanation of carbon that occurs during the H2pretreatment process.From Fig.4(d),the promoting activities of different promoters for FePc catalysts were in the order of K >Cs >Na ≈Li.The best catalytic performance was obtained for pyrolysis at 600 °C with 10% Cs doped and 700 °C with 3% K doped FePc.Two-dimensional (2D)FeOOH nanosheets encapsulated by mesoporous silica (mSiO2)FeOOH@mSiO2were prepared [26],as shown in Fig.4(e).Owing to the adhesion of the mSiO2coating and the limited space between them,the reduced Fe maintained its 2D morphology and exhibited high sintering resistance in the harsh Haber-Bosch process.A silica layer was successfully coated on FeOOH nanosheets.The distribution of Fe and Si in the coated samples was significantly more uniform than that in the supported samples.The ammonia production rate of the SiO2-coated FeOOH sample was 4.5 μmol∙g-1∙s-1at 425 °C,which was 50% more active than that of the SiO2-supported FeOOH sample (3.1 μmol∙g1∙s-1).Differential interactions of Fe with mSiO2and Al/mSiO2might induce another interfacial (metal-oxide) contact,thus changing the local Fe structure as well as the corresponding active sites.The catalysts containing two-dimensional Fe nanostructures (embedded in porous silica layers)were rich in surface step/kink sites.Compared to the catalysts containing Fe nanoparticles on silica spheres,the 2D Fe catalysts exhibited higher catalytic activity.Compared to the non-doped catalyst (Fe@mSiO2),the Al-doped mSiO2catalyst (Fe@Al/mSiO2) exhibited slightly lower activity and higher activation energy,indicating that the Al doping introduced structural and electronic effects,which negatively changed the nature of the active sites.Replacing part of the Fe with Co improved the activity of catalysts and positively affected the active sites(Fig.4(f)).Silicon dioxide was a better carrier than γ-Al2O3for both FeOOH nanosheets [55].
Embedded iron-based particles were likewise investigated.Liet al.[56] proposed computationally that Fe-N2complexes hosted on nanographene could facilitate N2fixation under ambient conditions.FeN3-graphene had a C3vsymmetric out-of-plane geometry with Fe atoms located outside the graphene plane and exposed at the top above the graphene layer,and the electron charge cloud being distributed near Fe,so Fe contributes the major spin moment.Because of its larger and more concentrated spin moment,the Fe site exhibited good N2adsorption and N2immobilization activity.N≡N bonds were first stretched by adsorption and then gradually elongated by hydrogenation until breakage.N2was trapped and could be either standing or recumbent in the formed complexes and was stable at room temperature,and the FeN3centre could be used in three ways to split the N≡N bond,each route being a 6-proton and 6-electron process.During subsequent hydrogenation,FeN3might participate in the reaction or acted only as a carrier for the transfer of electrons.And it was calculated that the energy required for the release of hydrazine during hydrogenation was not satisfied at room temperature.The stretching effect of the adsorption reaction was comparable to that of the hydrogenation reaction.The collaboration between graphene,as an electron reservoir,and FeN3,as an active site for N2immobilization and as a transporter for electron,assisted the system as a new catalyst for ammonia synthesis.
Simple iron-based catalysts can also be revitalized by alternative approaches.Hanet al.[57] achieved a final ammonia concentration of up to 82.5% (vol) better than the existing Haber process by using mechanochemical ball milling in a reaction,where the high defect density generatedin situby inexpensive untreated iron powder and the diverted energy from dynamic relaxation during severe impact accelerated ammonia production at mild conditions down to 45°C and 0.1 MPa.A series of unique properties were produced by the mechanochemical ball milling process in the iron catalyst used.Initially stable N2was attracted to the iron particle flaws and disintegrates into atomic N.Subsequently it was hydrogenated into NHx*species(x=1–3).Aided by extra transfer energy,the highly adsorbed NHx* species were then separated away from the Fe-surface.Eventually,the resulting ammonia is released.
3.2.Ru-based catalysts and other catalysts
Considering Sabatier principle,the best catalysts should bind atoms and molecules at moderate strength: neither too weak to activate the reactants,nor too strong to desorb the products.An obvious linear scale relationship exists between the transition state energy of N2dissociation in ammonia synthesis and the adsorption energy of nitrogen which is a variable that can be used independently to quantify bond energies on the surface of the metal concerned [58].The Fig.5(a) reveals the relationship between the catalytic activity of different catalysts and the adsorption energy of nitrogen.N adsorption energy is a surface property that responds to surface-mediated interactions that can be detected between transition metals and nitrogen [59].It is a research hotspot since Ru element is located near the top region of the volcanic shape curve,as shown in Fig.5(b).Ru-based catalysts allow milder operating conditions,such as lower synthesis pressures (7–10.5 MPa) and lower synthesis temperatures,while maintaining a higher conversion rate than conventional systems.Ru generally has to be paired with a support body[60].Ru precursors exists in a variety of forms.The most common ones are Ru3(-CO)12and RuCl3.In catalyst samples prepared from RuCl3,the particle size homogeneity of Ru is poor,while the residual Cl ions tightly bound to the metal surface or to the carrier adversely affect the catalytic behavior.Ammonia production rates for silica-loaded Ru cluster catalysts obtained from Ru3(CO)12are faster than those on catalysts prepared from RuCl3[61,62].The Ru3(CO)12clusters are adsorbed on the surface of active carriers such as alumina,the metal particles highly are dispersed and immobilized on the active sites of the carriers after decomposition and reduction to metal [63].However,it is not absolute.It has been recently found that the activity of catalysts prepared with Cl-containing precursors was much higher than that of catalysts prepared with non-Cl precursors when loaded with 1%(mass)Ru on MgFe2O4support(Fig.5(c)).The RuCl3precursor provided the highest Ru dispersion,while in the non-Cl precursors Ru particles were deposited in aggregates on the surface of the MgFe2O4carrier.Perhaps the high acidity of the aqueous solution leads to the rearrangement of MgFe2O4elements,which increases the concentration of Mg on the surface and forms Mg films.The coexistence of Mg films and Cl can effectively obtain a high dispersion of Ru particles,thus improving their catalytic activity [27].Besides,K2RuO4and Ru(NO)(NO3)3are also available as precursors.
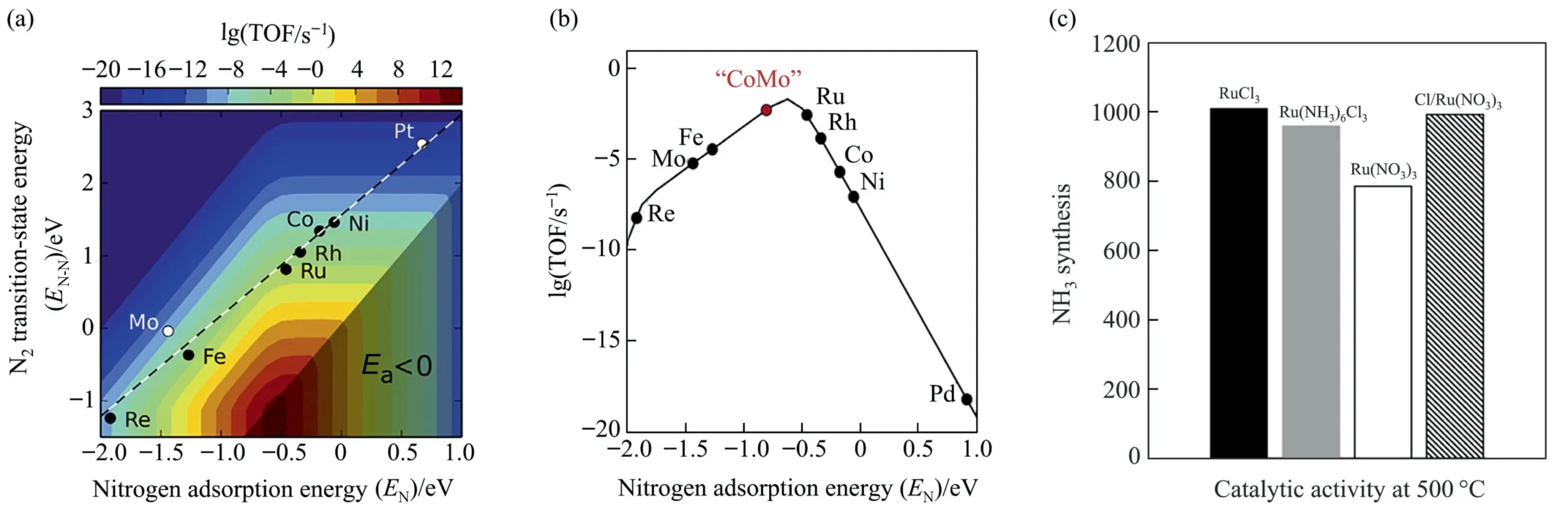
Fig.5. (a)Ammonia synthesis rates at FCC/HCP metal step sites and scale lines as a function of N2 adsorption energy and N2 dissociation potential.Reproduced from Ref.[59]with permission of Elsevier,copyright 2015 (TOF=turnover frequency).(b) Traditional volcano map.(c) Comparison of catalyst activity prepared using different precursor systems on MgFe2O4.Reproduced from Ref.[27] with permission of Chemistry Europe,copyright 2020.
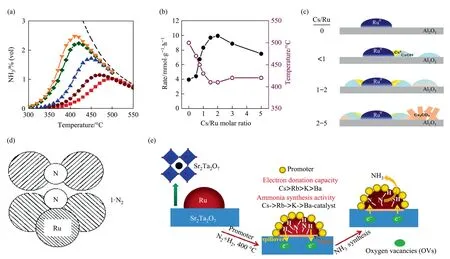
Fig.6. (a)Ammonia synthesis efficiency at different temperatures for different Cs/Ru ratio catalysts,The curves in order from bottom to top referring to Ru/γ-Al2O3,0.5Cs-Ru/γ-Al2O3,0.75Cs-Ru/γ-Al2O3,1.5Cs-Ru/γ-Al2O3 and 5Cs Cs-Ru/γ-Al2O3.(b)Relationship between Cs/Ru ratio and ammonia synthesis efficiency.(c)Existence of three types of Cs/Ru ratio carrier surface species.Reproduced from Ref.[28]with permission of Elsevier,copyright 2022.(d)B5 locus structure.Reproduced from Ref.[68]with permission of Wiley Online Library,copyright 2001.(e)Sketch of ammonia production from Sr2Ta2O7 with promoters.Reproduced from Ref.[46]with permission of Elsevier,copyright 2020.
Carriers/promoters of Ru crystals supported by recent researchers are categorized as diverse:Al2O3[28],perovskite type[29],lanthanide[31,32,64,65],zeolite[33,66,67],hydride/nitride,Ru/C12A7,etc.Al2O3is already used as a frequent carrier in industrial catalytic technology by virtue of its porous and robust framework nature.Recently,the catalytic properties of Cs-Ru/γ-Al2O3with Cs/Ru molar ratios of 0.25–5 were investigated (Fig.6(a)) [28].As illustrated in Fig.6(b),the best performance was centralized on 1.5Cs-Ru/γ-Al2O3,which yielded ammonia at 300 °C with a maximumrNH3of 9.7 mmol∙g-1∙h-1at 410 °C.5Cs-Ru/γ-Al2O3had the potential to catalyze low-pressure,intermittent ammonia synthesis reactions with high activity,good stability,and fast responsiveness.The catalyst demonstrated maximum ammonia precipitation efficiency at H2/N2molar ratio of 1 due to hydrogen poisoning.The specific surface area (SBET) value,gross value and porosity of the prepared Cs-Ru/γ-Al2O3catalyst gradually decreased with increasing Cs/Ru molar ratio.The Cs firstly remained in the acidic position of γ-Al2O3and then adhered to the Ru boundary,as the Cs/Ru molar ratio increased in the range of 0.5–1.5.Cs2CO3was formed at high Cs/Ru molar ratio(>1.5)(Fig.6(c)).During reductive activation,the reduced metal Ru particles and the reduced CsOH achieved a maximum at a Cs/Ru molar ratio of 1.5 and showed high ammonia synthesis activity.Density functional theory(DFT)calculations indicated that CsOH diminished hydrogen poisoning caused by hydrogen spillover and provided new active sites for ammonia synthesis.In addition,electron-promoting effects were observed through hydrogen spillover when reduced Cs species were formed at the interface of Ru and CsOH active sites.The Cs0/CsOH-Ru active sites of Ru/γ-Al2O3catalyst not only generated new active sites,but also changed the surface alkalinity/stability,facilitating the adsorption and desorption of H2,N2and ammonia,and promoting the ammonia synthesis process as well [28].
Chalcogenide oxide Sr2Ta2O7,as a carrier for Ru NPs,exhibits high alkalinity and high electrical conductivity.Huanget al.[29]synthesized 2% (mass) Sr2Ta2O7carriers by hydrothermal impregnation method.Excessive addition of Sr(OH)2to the hydrothermal synthesis speeded up the dissolution and nucleation rate of Ta2O5in pure-phase Sr2Ta2O7crystals,resulting in the formation of nanowires with smaller diameters.It was anticipated that the alkaline reduction carrier Sr2Ta2O7nanowires could suppress hydrogen poisoning on Ru surface by hydrogen spillover induced SMSI,release more B5 active sites,promote the dissociative adsorption of N2,enhance the number of N atoms on Ru surface as well as strengthen the activation of N2molecules in ammonia synthesis reaction,further promoting its reaction with H atoms and kinetic NHxformation.The so-called b5-type site consists of three Ru atoms on a layer on the Ru(0001) platform and two Ru atoms on the upper layer (Fig.6(d)) [68].
Alkali (earth) metals (Rb,K,Cs,and Ba) as promoters were equally examined.The outcomes demonstrated that the enhancements of promoter were in the following order: Cs >Rb >K >Ba(Fig.6(e)).The alkali metal promoters could boost the electron density on the Ru surface,thus enhancing the dissociation of N2through electron transfer.The four promoters exhibited various enhancements to the ammonia synthesis,with 5Cs-2Ru4STO(promoter to Ru molar ratio of 5,2% (mass) Ru raw material Sr(OH)2/Ta2O5molar ratio of 4) showing the highest performance.The appearance of Cs generated more OVs which along with the Cs promoter provided electrons to the Ru catalyst[29].The performance of known highly active ammonia catalysts could also be improved using unconventional promotion techniques.Shadravanet al.[30]usedin situpreparation techniques to promote Ru/C catalysts directly by metal Cs vapor in a high pressure plug flow reactor without any air exposure and with minimal oxygen impurities(especially O2and water).The higher activity of thein-situpromoted catalysts was not due only to the smaller particle size,on the contrary,the size ofin-situcatalysts was larger.An essential point in the stability performance ofin-situpromoted catalysts was the virtual absence of compounds containing O Cs2O,CsOH,CsN3,and CsH were readily reduced during the process to form adsorbed Cs*,which was the critical species that reduces the dissociation transition state energy of N2as a promoter,while all other species increased the activation energy.The electric field evoked by adsorbed Cs was associated with attractive interactions between dipoles induced by surface transition state N2molecules.An increase in the stability of the transition state with increasing Cs coverage led to an increase in the stability of the transition state,solving the problem that the rate of ammonia synthesis over nonin situcatalysts hardly varies with the ratio of Cs/Ru.The three H*atoms around the active site needed to be removed before dissociation of N2from the pristine Ru surface can occur.In contrast to the low Cs coverage model where two additional H atoms need to be removed,for the high Cs coverage model,there is also less impact on H2poisoning as only one H atom needs to be removed for the two Cs atoms.
Lanthanides are capable of being both promoters and supporters.One of the effects of being a promoter is to alleviate hydrogen poisoning,which means that hydrogen seems to adsorb more strongly on the surface of Ru atoms and covers almost all the Ru surface.The strong adsorption of hydrogen inhibits the activation process resulting in a delayed activation of nitrogen [64].La2O3is an active promoter of Ru/MgO.The activation of catalysts with lanthanide promoters is approximately twice that of the promoterfree sample M (Ru/MgO) [65].The Ru/CeO2catalysts with various surface oxygen vacancies on CeO2nanorods and nanotubes were plotted against each other (Fig.7(a)).The conversion of Ce4+to Ce3+and the formation of Ru—O—Ce bonds on the surface of CeO2nanorods contributed to the amount of oxygen vacancies.The low crystallinity and high concentration of oxygen vacancies of Ru species enhanced the adsorption of hydrogen and nitrogen,and also led to the desorption of surface hydrogen in the form of H2,thus exhibiting a higher ammonia synthesis activity.In contrast,lower catalytic activity was attributed to the presence of metallic Ru particles on the surface of CeO2nanotubes with large particle size and low concentration of oxygen vacancies and most of the consumed hydrogen material during the formation of water[69].Recently,a combination of Ce and La elements(Ru/La0.5Ce0.5-O1.75) has been reported.This was a catalyst consisting of La0.5-Ce0.5O1.75solid solution: Ce and La were uniformly dispersed in the oxide carrier,and Ru particles are strongly anchored on La0.5-Ce0.5O1.75synthesized from CeO2and La2O3.As shown in Fig.7(b),it exhibited a high rate of ammonia synthesis(31.3 mmol∙g-1∙h-1) due to the intense interaction between many Ru active sites and the reduction carrier.The formation of solid solution complex oxides disturbed the adsorption of La2O3to water and the crystal growth of the oxidation carrier.Due to a result of the reduction of Ce4+to Ce3+and the formation of oxygen vacancies,the partially reduced carriers covered part of the Ru particles leading to a greatly enhanced electron transfer.The electron transfer to the antibound p orbital of N2,weaking the N≡N triple bond.The high pre-reduction temperature induced SMSI and promoted Ru site activity,but reduced the number of Ru active sites,which together resulted in an optimal pre-reduction temperature of 650°C,significantly higher than the reaction temperature of Ru-catalyzed ammonia synthesis.The Ru/La0.5Ce0.5O1.75,a solid solution homogeneously dissolved in a cubic elongate structure,had the optimal catalytic performance (Fig.7(c)) [31].
Combination of two or more metal oxides as vehicles are thought to yield diverse functionalities from each oxide,allowing new properties in catalysts.Mg and Er are regarded as common vehicles because of identical dispersion.Lanthanide oxides typically have lower surface area but higher catalytic ammonia synthesis activity than MgO,and mixtures of the two can better support Ru-based catalysts by obtaining higher surface area from MgO as well as electron donor promotion from lanthanide.1%Ru mixed oxide carriers of MgO-Er2O3with various molar ratios of Mg/Er were fabricated by co-precipitation method and the NH3efficiency varied with the value of Mg/Er ratio(Fig.7(d)).The excess Er2O3at Mg/Er ratio of 1/5 increased the crystallinity of MgO inhibiting the crystal growth and the MgO fraction remained in the amorphous or fine-grained state.The surface area grew with the content of MgO,and afterwards a more stable state was reached.A further increase in MgO content caused substantial structural changes,leading to a decrease in surface area and high growth inhibition of Er2O3crystal.The ammonia synthesis activity of Ru/MgO-Er2O3accompanied by a well dispersed Mg/Er molar ratio of 25/1 was comparable to that of pure Er2O3supported Ru catalysts,which somewhat mitigated the problem of expensive production cost [32].
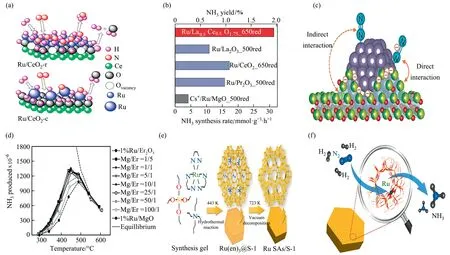
Fig.7. (a) Different surface oxygen vacancies of Ru/CeO2 nanorods compared with nanotubes.Reproduced from Ref.[69] with permission of American Chemical Society,copyright 2018.(b)Ru/La0.5Ce0.5O1.75 Optimal ammonia synthesis efficiency.(c)Probable activation mechanism of N2 on Ru/Ce0.5La0.51.75_650 red.The gray ball is for Ru,the red ball is for oxygen atoms,and the two-color ball is for La/Ce.Reproduced from Ref.[31] with permission of Royal Society of Chemistry,copyright 2018.(d) The activity tendency of various Mg/Er ratio Ru catalysts produced with MgO-Er2O3 medium.Reproduced from Ref.[32] with permission of Elsevier,copyright 2020.(e) Monatomic Ru acquisition path.(f) RuSAs/S-1 simplified ammonia production process.Reproduced from Ref.[33] with permission of American Chemical Society,copyright 2019.
Since Ru planes are independent of N2dissociation,it is necessary to select suitable carriers and promoters as much as possible to make small Ru clusters reach the same active state as the bulk Ru metal.Zeolites are favored carriers for Ru metal.The dimensions of the pores or the cages of the zeolite constrain the particle size within the crystal and the basicity of the zeolite can be changed by cation alteration [66].Cisneros and Lunsford [67] also discovered that the activity of zeolite-supported Ru was firmly anchored by the presence of cations in the zeolite-the more basic the zeolite,the more active the catalyst.The intense basicity of the added promoter induced localized destruction of the zeolite framework.Zeolites also supplied acid-related active centers,which hindered the mechanistic elaboration of the sites.Pure silica zeolite-loaded Ru single-atom catalysts were employed to overcome this difficulty.As shown in Fig.7(e),single Ru atoms were obtained from [Ru-(NH2CH2NH2)3]Cl3,where Ru3+ions were latched by three glycoside ligands.The acquired samples had a nanoscale hexagonal prismatic morphology.According to some electron microscopic analyses,Ru atoms were distributed uniformly in the crystals of zeolite,and there were no subnanoclusters inside the zeolite crystal or outside the crystal surface.The induced Ru complexes resided in the zeolite channels rather than causing lattice defects.Suitable atmosphere,heat treatment temperature and Ru loading were critical factors for the successful preparation of single-atom catalyst (SAC).The catalytic activities of RuSAs/S-1 were all higher than those of Cs-Ru/MgO.The ammonia synthesis rate of Ba-RuSAs/S-1 obtained after the addition of promoter Ba ions reached 1389.5 μmol∙g-1∙h-1at 400 °C,which was approximately 2 orders of magnitude higher than that of the unpromoted catalyst.The oxidation state of a single Ru atom approaches that of RuO2,and the higher oxidation state led to a lower d-band center,which provided less electron feed back to the antibonding orbital of the N2molecule and less electrons to the H2molecule,inhibiting the H-poisoning phenomenon.N2* was adsorbed on the exposed Ru single atom position linearly.It had weaker affinity for the Ru δ+center compared to N2*,thus facilitating the physisorption of H2within the zeolite channel.The rate-determining step was the reaction of physiosorbed H2and adsorbed N2*,which yielded two ammonia molecules with simultaneous structure A release,completing the cycle.RuSAs/S-1 simplified ammonia production process is shown in Fig.7(f) [33].
Ru-loaded titanium carbide nitride (TiCN) was synthesized by ball milling.Ru/3TiCN/ZrH2(3% (mass) Ru) catalyst was synthesized at 400 °C and 1 MPa with very low methane concentration in the side reaction[34].The incorporation of Ru—N bond between Ru metal and TiCN could successfully restrain the agglomeration of Ru particles and create more B5 sites,which promoted the transfer of electrons from Ru position to the antibonding orbital of N2.With the assistance of Ru,N vacancies on TiCN were prone to form*NNH by N2hydrogenation reaction which follows bonding mechanism.The generation energy of N vacancies in TiN was lower than that in TiCN (Fig.8(a)).The reversible hydrogen storage capacity of hydride(ZrH2)could effectively inhibit hydrogen poisoning.Unlike conventional Ru-based catalysts which usually follow dissociation mechanism,Ru/3TiCN/ZrH2followed integrated dissociation and binding mechanism,giving the reason of the superior ammonia synthesis rate of our catalysts under mild conditions(Fig.8(b),(c)).
Although valid promoters can be alkali/alkaline-earth metal hydrides or nitrides that confer sufficient electrons to Ru or stimulate electrostatic effects of Ru surface reactants to enhance catalytic activity,the possibility of forming metal amides by virtue of their own chemical activity in reaction with the resulting ammonia can cause instability [2,35].Ru-loaded charged metallic group [Ca24Al28O64]41(e–)4(C12A7),a crystalline cavity that captures electrons in the anion,has been discovered in recent years[70].The unit cell of the positively charged framework structure C12A7consists of 12 sub-nanometer sized cages,four of which contain four O2–ions as counter anions,connected with a monoxide layer to achieve electrical neutrality.The chemical reduction process compensates for the positive charge on the cage walls by extracting two O2–ions from the 12 cages and injecting four electrons into the two O2–ions in the cavities (Fig.8(d)) [70–72].The electrons encapsulated in the cage can easily form H–by heating in H2gas.The H–ion desorbs as an H2molecule at about 400 °C,leaving the electrons in the positively charged framework of C12A7.The hydrogen atoms on Ru can simply escape into the negatively charged framework of C12A7,thus preventing the hydrogen atoms from occupying the Ru surface while the electrons are transferred to Ru.The increase in the Fermi energy level of the metal leads to a significant decrease in the work function of Ru,thus allowing the H–ion to react with the N formed by the dissociation of N2to form NH3(Fig.8(e)),and the electrons wrapped in the negatively charged framework of C12A7are used repeatedly during the reaction [35,73].
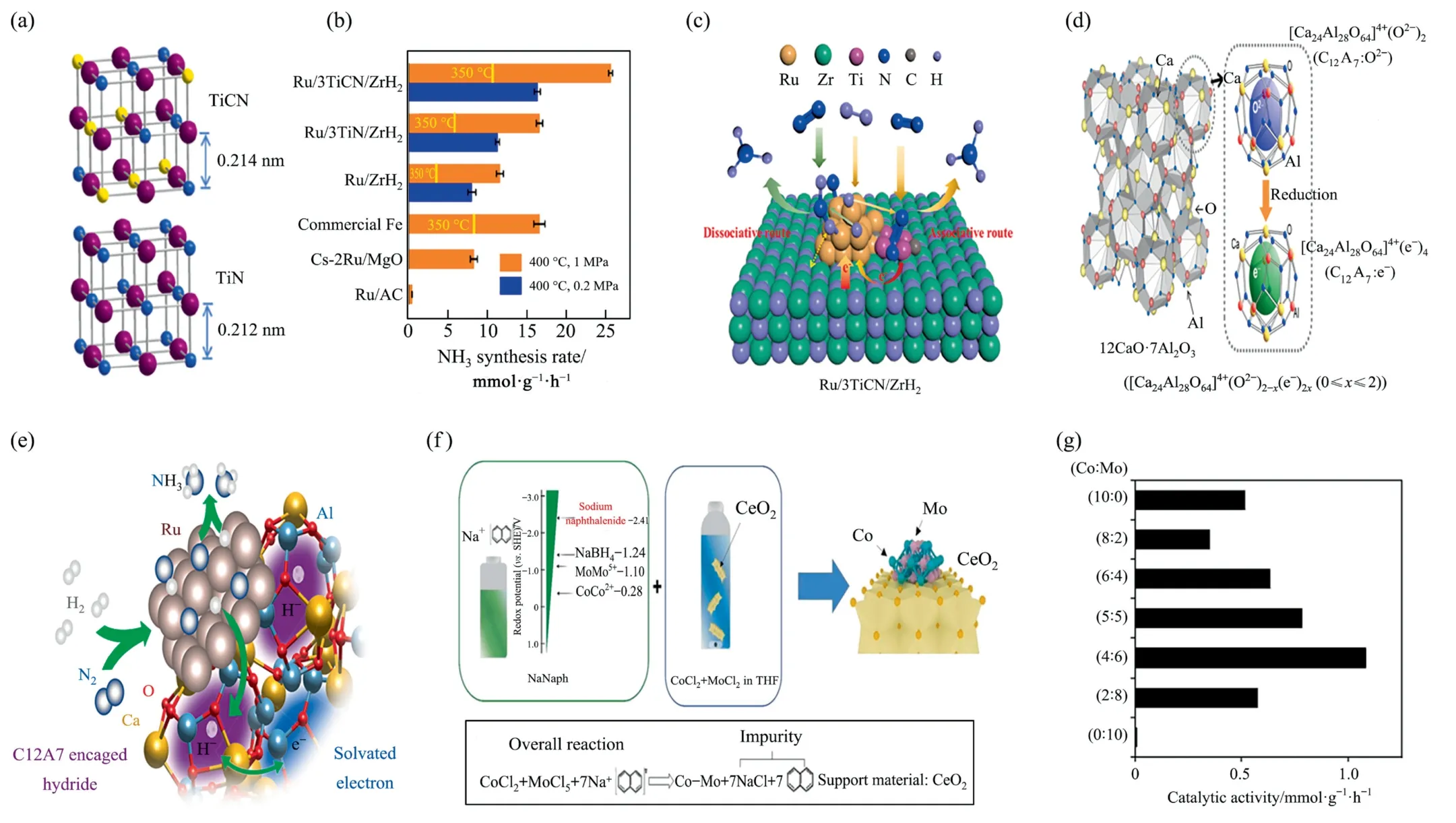
Fig.8. (a)TiCN and TiN cell structure.(b)NH3 synthesis performance of Ru/3TiCN/ZrH2 and each control group at 400°C under 0.2 or 1 MPa.(c)Structure of Ru/3TiCN/ZrH2.Reproduced from Ref.[34] with permission of American Chemical Society,copyright 2022.(d) Framework structure of each part of C12A7.Reproduced from Ref.[70] with permission of American Chemical Society,copyright 2017.(e) Schematic diagram of Ru/C12A7 reaction.Reproduced from Ref.[71] with permission of American Chemical Society,copyright 2017.(f)Schematic diagram of the synthesis of Co-Mo(X:Y)/CeO2 catalyst.(g)Catalytic activity of various Co/Mo ratio catalysts for ammonia synthesis at 0.1 MPa and 400 °C.Reproduced from Ref.[9] with permission of Elsevier,copyright 2018.
However,the high cost of catalysts associated with Ru (noble metal) is a major drawback that has inspired the development of inexpensive non-precious metal catalysts.Combining metals on both sides of the volcano diagram,such as Co and Mo,or Ru/Co is an effective stage to achieve the desired interactions of intermediate N.Tsujiet al.[9]introduced CeO2into a tetrahydrofuran solution containing cobalt chloride and MoCl5in multiple steps to obtain Co-Mo/CeO2.Co and Mo were present in close proximity and synergistically interacted(Fig.8(f)).The authors identified that when Co/Mo equalled 4:6,the ammonia synthesis rate would reach a maximum value (1.08 mmol∙g-1∙h-1) shown in Fig.8(g).The reaction mechanism of ammonia synthesis was comparable to that of Co3Mo3N.N vacancies were formed more readily over Co-Mo/CeO2catalysts than over bulk Co3Mo3N.The N2dissociation step on Co3Mo3N nanoparticles was enhanced by the electronic contribution supported by reduced CeO2.A suitable proportion of Cr and K,serving as promoter,led to a well-developed porous structure.The total surface area and catalytic activity were increased by more than 50% compared with non-promoted catalysts [74].Ru atoms were inserted to form RuCo double monoatomic active sites on the surface layer where N of g-C3N4coordinates with Co.The developed RuCo dual single-atom catalyst(RuCoDSAC) had an ammonia synthesis rate of 1.24 mmol∙g-1∙h-1at 200 °C.The RuCoDSAC structure could effectively foster the transfer of electrons from pyrrole N to Ru atoms on the surface to promote adsorption,activation and hydrogenation of N2.N2adsorbed at the Co center to form hydrides was transferred to the Ru site to react with the activated N2to generate N2Hxintermediates.Meanwhile surface intermediates such as *N2Hxand *NH3were readily desorbed on RuCoDSAC with narrow d-band centers[36].
Ru nanoparticles(NPs)have been proven to be their active sites.Ru catalysts with large nanoparticles subject to over-adsorption on the H2surface,blocking N2activation.As per the theoretical model-the Wulff structure suggests that with the size of Ru entities due to the lack of active sites,the sub-nanoclusters should have little activity.However,it has been shown that narrowing down to sub-nanoprecious metal clusters,atomic clusters or even single atoms is an effective strategy to enhance ammonia synthesis.N2activation pathways change as clusters,single atoms fully expose the catalytically active edge or corner sites and generally exhibit significantly enhanced properties.By adjusting the precursors and/or loading,single atom,atomic clusters,and sub-nanocluster ruthenium catalysts are synthesized by breaking the limits of nanoparticles.The sub-nanocluster Ru catalysts not only exhibit different properties from the NPs,but also have different activation pathways for N2.The intense intracluster interactions of the Ru clusters enable the Ru d orbitals to form strong interactions with the s and p orbitals of the N2molecule,causing activation to occur more readily on the Ru ACC or sub-nanocluster than on the Ru NPs[75].The result is a weakening of the N2bond orbital.The adsorption,activation and hydrogenation of N2by the catalyst is significantly facilitated by the mechanism of association with a very small reaction energy potential barrier [76].
To prevent the polymerization of small size Ru,various approaches can be employed.Zhouet al.[37] employed the colloidal particle impregnation method to deposit colloidal Ru nanoparticles on BaCeO3carriers.The decrease in Ru size enhances the production of Ce3+and O vacancies in BaCeO3,thus providing electrons to the Ru centre,and subsequently the Ru 4d orbital gave electrons to the antibonding orbital of the N2molecule to reduce the N2activation potential barrier and also enhances hydrogen spillover from Ru to BaCeO3to relieve hydrogen poisoning for efficient ammonia synthesis.To reduce the particle size distribution of the catalyst,Liet al.[76] used a carrier with copious porous channels and cavities like MOF (MIL-101),superior to zeolites,to constrain the metal cluster size and anchor the reaction sites.Through a dual fit strategy of metal-linked porous materials,the metal-encapsulated MOF hosts were pyrolyzed after the addition of MgO or/and Cs2O,and the embedded metal clusters were eventually trapped in the shrinkage cavities.
3.3.Metal nitride catalyst
Rare earth metal nitrides such as CeN and LaN have received a lot of attention due to their nitrogen vacancy activation for ammonia synthesis,the nitrogen vacancy formation energy (NVFE) governs the catalytic performance and is a general rule for the design of nitride-based catalysts[38].The presence of N vacancies in CeN could activate both N2and H2during the reaction and its catalytic performance is much higher than other reported unloaded catalysts for ammonia synthesis.One of the attractions of LaN/Ni was its low NVFE.The reaction of the lattice N of LaN directly with dissociated hydrogen (H*) on the Ni surface yielded ammonia and N vacancies.where the N2molecule was subsequently adsorbed and activated at the N vacancy site and continuously reacts with H* to achieve a stable catalytic cycle.In the condition of a nitride loaded with Ni,H2and N2were activated at the Ni metal and VN sites individually.Without Ni loading,the VN site can activate both H2and N2to stabilise ammonia production.Yeet al.[39] also reported a synergistic mechanism for the binding and dissociation of Co/CeN in the synthesis of ammonia over catalysts under mild reaction conditions.The formation of N vacancies in CeN also promoted the efficient cleavage of N2on Co.
The finitely charged hydrogen atom in lithium hydride acting as a strong reducing agent eliminates the activated N atom from the transition metals (TM) or its nitride (TMN) and serves as a direct source of hydrogen which bonds with the N atom to form Li2NH/LiNH2to further heterolyze splitting H2,releasing ammonia and regenerating the lithium hydride (Fig.9(a)).Co-creation between TM(or TMN)and lithium hydride could usefully separate Nadfrom TM(N)to free TM site and then recycled the reaction[40].The combination of Co with BaH2formed the catalyst which had low temperature reaction activity (Fig.9(b)) [42].The nickel nitride with embedded nickel also obviated the common dual site mechanism of proportional relationship (Fig.9(c)) [41].In addition,the insertion of other substances into the hydride also allowed to obtain excellent ammonia production efficiency.As shown in Fig.9(d),ammonia productivity of KH0.19C24obtained by intercalating KH into the layers of the graphite host material at 250–400 °C and 1 MPa,was comparable to that of the classical noble metal catalyst Ru/MgO and the synthesis principle inclined to comply with the associative alternating pathway [8].
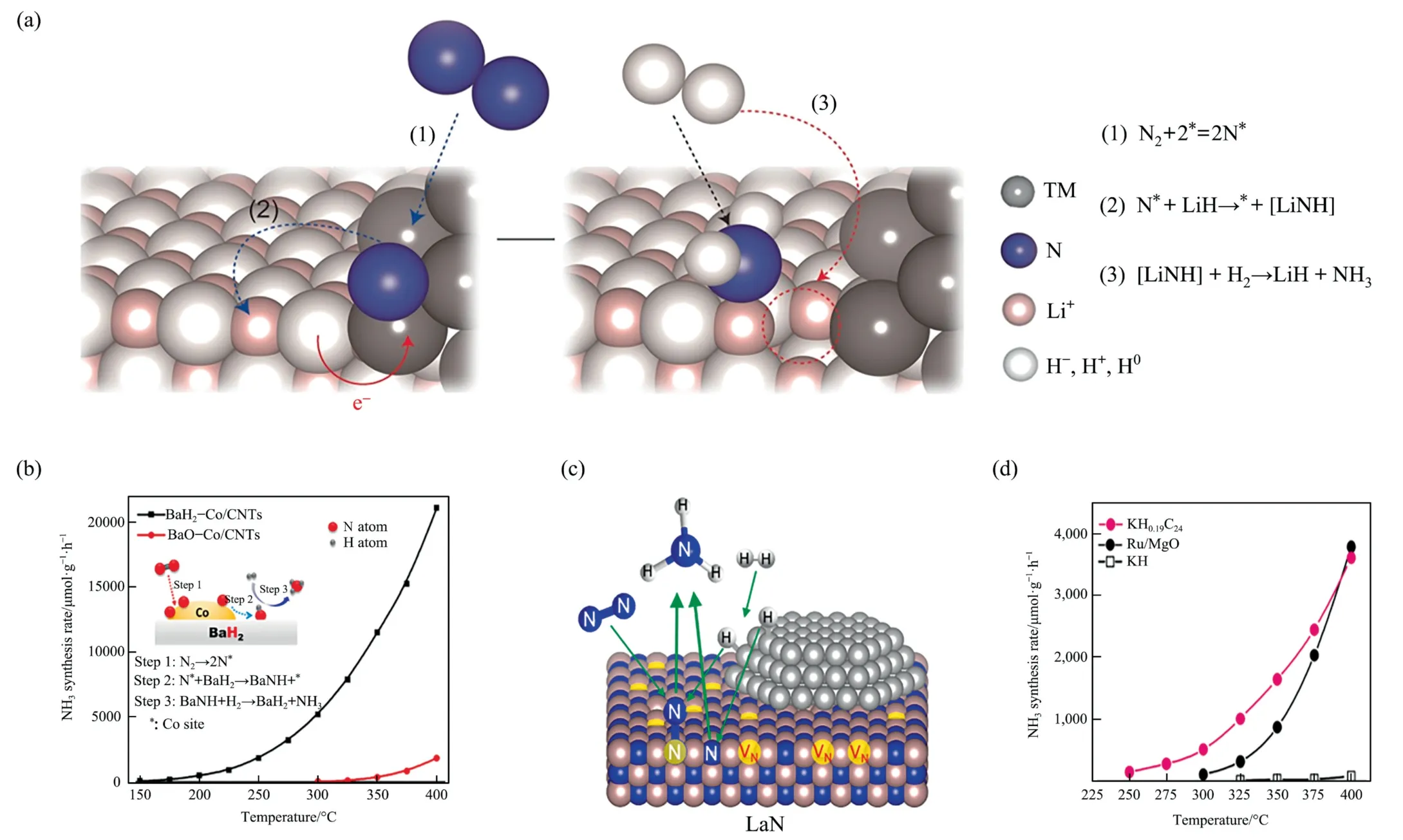
Fig.9. (a) Reaction mechanism of ammonia synthesis over TM(N)-LiH catalyst.The white balls represent H0,H– and H+,the basketball represents N,the powder ball represents Li+ and the gray ball represents TM.Reproduced from Ref.[40] with permission of Nature Publishing Group,copyright 2016.(b) Mechanism and efficiency of ammonia synthesis reaction over BaH2-Co/CNT catalyst.Reproduced from Ref.[42] with permission of Nature Publishing Group,copyright 2020.(c) Structure of ammonia synthesis over loaded transition metal catalysts (TM/LaN).Reproduced from Ref.[41] with permission of American Chemical Society,copyright 2017.(d) KH0.19C24 corresponds to the equivalent ammonia productivity of Ru/MgO at 250–400°C and 1 MPa.Reproduced from Ref.[8]with permission of Nature Publishing Group,copyright 2020.
4.Electrocatalytic Synthesis of Ammonia
Electrocatalytic ammonia synthesis,an alternative to thermal catalysis,is one of the most widely used technologies,which provides a pathway to zero CO2emissions by greatly reducing the carbon footprint of the chemical industry due to its ability to produce ammonia from pollutant-free water at lower temperatures and pressures (Fig.10(a),(b)).Recently,lots of efforts have been made to develop electrochemical ammonia synthesis processes.The advantages of simple equipment and moderate efficiency permit the construction of electrochemical processes from small distributed facilities to large production plants.The high energy density,relatively abundant hydrogen content and ease of compression into liquid form for storage have also motivated the use of ammonia instead of hydrogen by electrolysis to address the current challenges in the context of today’s energy crisis[2,3,77,80,81].The electrocatalytic nitrogen reduction process on multiphase surfaces is inclined to follow a combination mechanism.More specifically,depending on the type of nitrogen adsorption and the order of hydrogenation,the combination mechanism can take place in three possible pathways: distal,alternating and enzymatic [11].The enzymatic pathway,which is the exact opposite of the Haber process in which the nitrogen adsorbed molecule is a lateral ligand model.Under these possible mechanisms,the reduction of nitrogen may lead to different products,including ammonia and hydrazine.
In general,there are four key points that make electrochemical ammonia synthesis difficult: (1) most of the catalysts explored have limited ability to bind and adsorb N2leading to unsatisfactory activation of N2in the first step of the reaction [82];(2) some strong binding and adsorption catalysts work well in the first step but produce intermediates that are difficult to protonate or cause the disappearance of ammonia;(3)electrocatalytic NRR in aqueous solution also has a strong competing side reaction,the hydrogen reaction (HER),which leads to low Faraday efficiency (FE) [83];(4)The ability of some metals to suppress HER is inconsistent with the ability to improve NRR.In principle,the electrocatalytic performance of NRR catalysts is characterized by three activity indicators: yield,FE and catalytic stability.The first two can be simply calculated by measuring physical quantities,and the last is obtained by time-dependent electrolysis tests[11].The electrocatalytic nitrogen reduction reaction(ENRR)has two additional mechanisms in parallel with the three previously mentioned in this review: the Mars-van-Klavern (MvK) process and Li making intermediary mechanism (Fig.10(c),(d)).In the MvK process,the crystalline lattice nitrogen atoms in the nitride form ammonia by hydrogenation,and the produced vacancies of nitrogen are then replenished by N2[78,83].During the lithium making intermediary mechanism,N2gets activated by reaction with lithium metal initially and then form lithium nitride,which can occur automatically in environmental conditions.Next,lithium nitride is protonated to form Limonium salt as well as ammonia.Ultimately,the Limonium salt is galvanically converted back to lithium metal,thus ending the cycle [79].

Fig.10. (a),(b) Flow chart of H-B process and electrochemical process for ammonia synthesis.Reproduced from Ref.[77] with permission of Royal Society of Chemistry,copyright 2021.(c)Mechanism of reactivity(right)and deactivation(left)of VN0.7O0.45.Reproduced from Ref.[78]with permission of American Chemical Society,copyright 2018.(d) Li-mediated mechanism.Reproduced from Ref.[79] with permission of Nature Publishing Group,copyright 2020.
Originating from the production of N2from nitrate,it is found that some catalysts are more selective for the generation of ammonia.Despite the need for three more electrons to convert nitrate to ammonia and the competition of N≡N bonds and other reactions like the formation of H2NOH,adsorbed N2O can be dissociated before conversion to N2at noble metal electrodes,skipping the generation of N≡N bonds,rendering nitrate-based electrocatalysis equally favored.Nevertheless,a variety of stable intermediates and products are implicated in the reaction process,such as nitrite,hydrazine,hydroxylamine,nitric oxide,nitrous oxide [84,85].
Galvanic NRR catalysts have various categorization methods following different classification criteria.For example,they can be classified by elements into several groups such as noble metals(Au,Ru,Rh,Pd,etc.),non-noble metals,and non-metals(including various composite materials).According to the operations performed,they can be further classified into types such as structural optimization,fabrication of vacancies,atomic doping,and nonstatic changes.This paper classifies the catalysts according to the required elements,summarizes the scientific research results that have emerged in recent years,and clarifies the development trend of electrochemical NRR catalysts.Table 2,Table 3 and Table 4 provide the summary of the performance of several relatively newly reported NRR processes.
4.1.Noble metal catalysts
Precious metals typically refer to platinum cluster metals as well as two monetary metals (gold,silver) and other eight metal elements,they possess strong chemical stability and hardly chemically react with other chemicals under general conditions[81,83].Au is the part of the optimal catalyst for electrochemical NRR[127],nevertheless,the limited availability and affordability of gold are serious obstacles to its massive industrial utilization.Construct proper morphology of Au-based catalyst is considered as a valid effective method for remarkable enhancement of electrocatalytic performance.
Inspired by activity of atomic dispersion catalyst [128],Wanget al.[86] added HAuCl4-4H2O aqueous solution dropwise into g-C3N4aqueous dispersion,an actual packing of 3.4% of atomically dispersed Au1/C3N4was obtained after a series of operations.As shown in Fig.11(a),the authors assembled a total electrolytic cell using platinum foil as the anode,Au1/C3N4as the cathode,the Ag/AgCl electrode as the reference electrode.A cation exchangemembrane separated the sulfuric acid aqueous solution electrolyte.Carbon fiber paper electrodes were loaded on both sides of the electrodes by means of a homogeneous ink acquired by distributing the prototype and the Nafion liquid with a volume ratio of 1:1.The gold atomic valence of 1 was studied by Extended X-ray absorption fine structure method and X-ray photoelectron spectroscopy (XPS).Au1/C3N4electrochemically reduced N2to NH4+under environmental circumstances with FE of 11.1% and the NH+4yield reaching 1305 μg∙mg-1∙h-1(approximately 22.5 times higher than Au NPs/C3N4),no hydrazine was detected in both the experimental and control groups.The calculations indicated that Au1/C3N4favored the pathway of N2reduction to ammonia by complex alternating catalysis (Fig.11(b)).Bader charge-buccal analysis affirmed that there was an appreciable charge removal from the gold atom to g-C3N4,causing the Au atom to generate a positive charge with 0.56 |e|.The electron exhaustion of the gold atom might relocate its d-orbit placement toward the Fermi energy level,thus strengthening its interaction with intermediates (e.g.,NNH*) and resulting in better NRR performance.Nazemiet al.[87] adjusted the peak localized surface plasmon resonance (LSPR) of hollow gold nanocages (AuHNCs) to regulate the dimensions and density of the pores.Hollow gold nanocages,who’s peak LSPR spectra shifted to the desired values of 635,715,or 795 nm,were prepared by the deionized water synthesis method that injecting HAuCl4into AgNCs solutions under vigorous stirring.AuHNCs-715 featured the maximum output of ammonia at–0.5 V(4.22 μg∙cm-2∙h-1)and the highest FE at–0.4 V (35.9%),which was a trade-off between greater amperage density and enhanced selectivity for HER (Fig.10(c)).As the position of the LSPR peak shifted from 635 to 795 nm,the gold concentration (% (mass)) in the microparticles grew from 33.0 to 64.7,accompanied by an augment in aperture size.The contradiction between the highest Au concentration and the lowest Au electrochemical surface area concentration appeared in AuHNCs-795.Excessive pore size obviously reduced surface area,however the existence of Ag lying in the hollow cavity of AuHNCs with smaller pore diameter (Ag capable of enhancing the evolution of H2)lowered the selectivity of electro-catalyst to NRR.Bismuth telluride with excellent micromorphology and surface attraction properties is a more suitable vehicle for synergists [129].Two-dimensional(2D)heterojunction Au-Bi2Te3nanosheets(AuBi2Te3NSs)are made up of bismuth telluride nanosheets with gold nanoparticles sedimented on[88].Because of the promising dispersion of Au nanoparticles and the outstanding cooperative effect of heterojunction composites,Au-Bi2Te3NSs showed sound nitrogen reduction reaction(NRR)performance in the ambient.The abbreviated synthesis method is given in the Fig.11(d).The dispersion solution prepared by mixing the catalyst with other solutions was applied uniformly on clean carbon writing paper and tested with carbon rods as counter electrode,Ag/AgCl served as reference electrode.The NH3yield of Au-Bi2Te3NSs was 32.73 μg∙mg-1∙h-1with FE of 20.39%at–0.4 V in 0.1 mol∙L–1sodium sulfate electrolyte (N2flooded) (Fig.11(e)).The reason why modulating the concentration of HAuCl4solution could change the gold content of the sample was attributed to that during the synthesis process,bismuth telluride and AuClcould undergo oxidation reactions resulting in the generation of electrons,which could be spontaneously transferred from bismuth telluride to AuCl,leading to the nucleation and growth of Au,forming Au nanoparticles.The characterization proved the hexagonal structure of Au-Bi2Te3NSs,with Bi and Te elements uniformly distributed throughout the nanosheets,while AuNPs were anchored on the surface of bismuth telluride NSs (Fig.11(f)).Tannic acid(TA)is an eco-friendly natural polyphenolic with many oxygenated phenolic hydroxyl functional groups on its rigid aromatic ring backbone,which is harder to rotate so that it can be used to obviate the agglomeration of catalysts.Hence,TA was pursued as a stimulator for precious metal nano-materials [130].The contribution of its structure to the electrochemical activity was demonstrated by the whopping 14.83%FE at–0.3 V with ammonia yield(15.71 μg∙mg-1-∙h-1) (Fig.11(g)) [89].TA-gold nanowires (AuNWs) were established by superficial amendment of AuNWs using oxygenenriched TA,but still retained the homogeneous nanowire structure of AuNWs(Fig.11(h)).The superior NRR reactivity and flexibility of TA-AuNWs could be ascribed to the unidimensional configuration of active Au,and the superficial amendment of oxygen-enriched TA.The hyperfine single dimension composition could deliver adequate reactive sites for NRR adsorption and activation of N2.In inclusion,the aerobic-rich TA lamination has lucrative interfacial adsorption locus,which enhances the reaction kinetics of NRR by increasing the probability of inter-collisions of the reaction species with the active site of the accelerator.

Table2 A summarization of some newly developed noble metal electrocatalysts for NRR

Table4 A summarization of some newly developed non-metallic and three-component composite electrocatalysts for ammonia synthesis
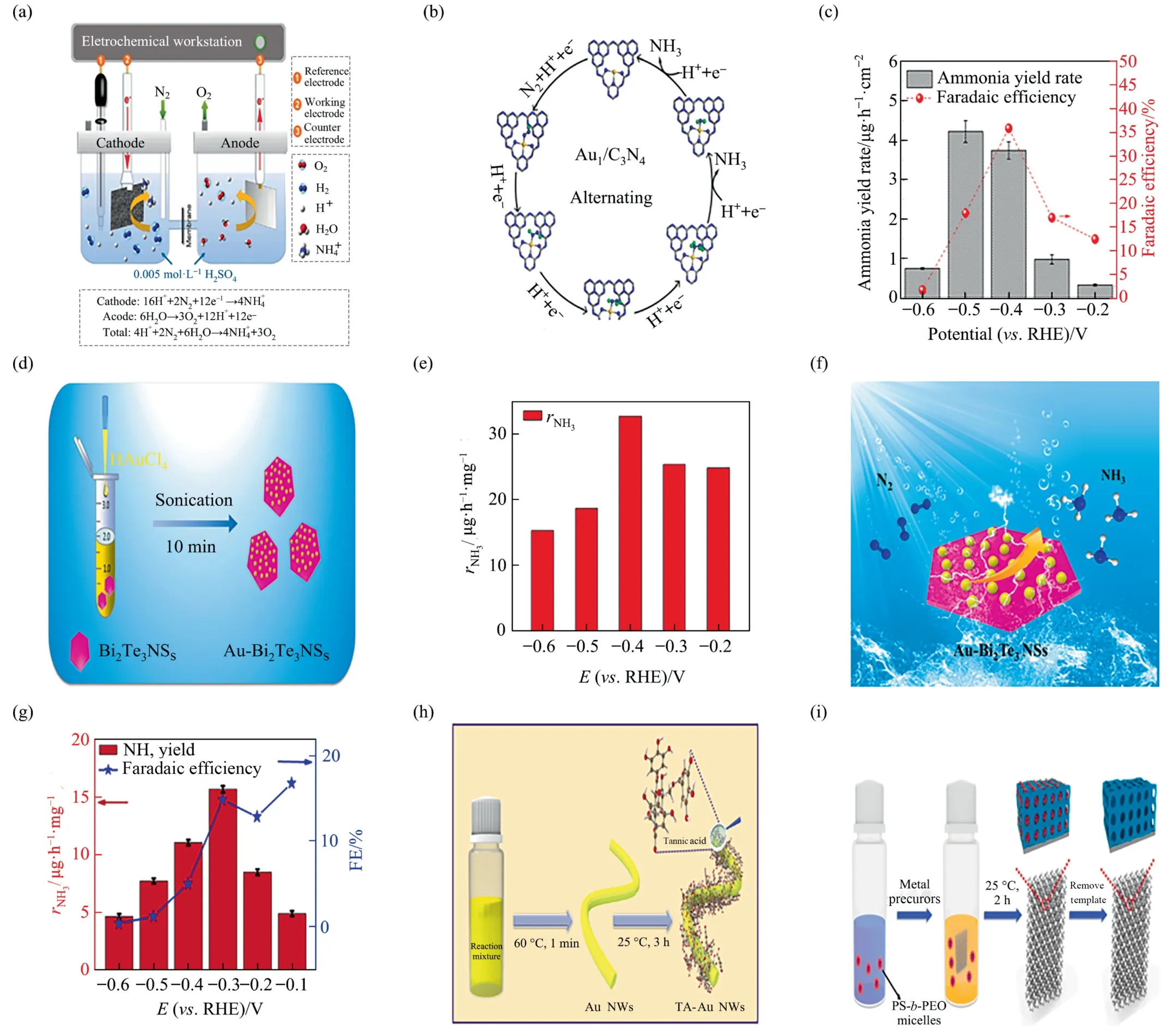
Fig.11. (a) Schematic diagram of electrolier for N2 electro-reduction experiment.(b) Mechanism of alternating cycles on Au1/C3N4.Reproduced from Ref.[86] with permission of Elsevier,copyright 2018.(c) Ammonia production rates and FE of AuHNCs in 0.5mol∙L-1 LiClO4 aqueous solution at various potentials.Reproduced from Ref.[87] with permission of American Chemical Society,copyright 2018.(d) Synthesis pathway of AuBi2Te3NSs.(e) Ammonia yields over Au-Bi2Te3NSs catalysts at diverse potentials.(f)Reaction path of AuBi2Te3NSs as catalyst.Reproduced from Ref.[88]with permission of American Chemical Society,copyright 2021.(g)Comparison of FE and ammonia production at different potentials.(h)Synthesis steps of TA-AuNWs.Reproduced from Ref.[89]with permission of Elsevier,copyright 2021.(i)Schematic diagram for preparing the mAu3Rh/NF.Reproduced from Ref.[90] with permission of Elsevier,copyright 2021.
The conventional methods employed for the preparation of porous precious metals typically cannot control the dimensions of the pore.The micelle assembly method has been proven to produce porous metals directly in the medium of aqueous solutions [131],which has prompted Yuet al.[91]used micelle assembly to bypass the mesoporous Pt-based material (polymer binder resulting in low activity)to synthesize self-supported mesoporous Au3Pd films(mAu3Pd/NF)on nickel-based bubble compounds with polystyrene PS-b-PEO.HAuCl4aqueous solution and K3RhCl6aqueous solution as raw materials [131],where PS-b-PEO was used as a stomatal reformer and Ni foam was used as both a primer and a reductant.mAu3Pd/NF exhibited prominent NRR characterization in 0.1 mol∙L–1sodium sulfate with NH3yield of 24.02 μg∙mg-1∙h-1and FE of 18.16% at–0.1 V.Transmission electron microscope(TEM) and energy dispersive X-Ray spectroscopy (EDX) revealed that the resulting product is the combination of gold and palladium,and XPS measurements implied the existence of electron transfer between Au and Pd elements,giving rise to a strong electron effect.The spherical micelles of PS-b-PEO were the key contributor to the generation of mesopores.Along with the reaction,Pd and Au ions were progressively reduced to formulate AuPd alloys,in which mesopores were shaped.The uniform depth increment of film was accompanied with the increase of the synthesis reaction time.In comparison with samples of different Au/Pd ratios,the output of ammonia and selectivity of mAu3Pd/NF were better than mAu/NF and mPd/NF,even over 3 folds than that of no-porous Au3Pd/NF.The well-balanced mesoporous nanostructures provided rich active sites,superb specific surface area and barrier free delivery paths.The result of theoretical calculation showed that the high activity was attributed to the effects of the formation of mesopore direct inducting by PS-b-PEO micelles,AuRh bimetallic composition effect and electronic effect as well as the synergistic effect of self-loaded mesoporous membrane structures.As shown in Fig.11(i),Wanget al.[90] has also proposed thein situsynthesis of mesoporous Au3Rh membrane on Ni foam material (mAu3Rh/NF).Thanks to the bimetallic mesoporous structure,the FE of mAu3Rh/NF is 23.84%and the ammonia yield was 26.29 μg∙mg-1∙h-1in 0.1 mol∙L–1sodium sulfate at–0.1 V,which was superior to other NRRs catalyst with different Au/Rh ratios.Ruthenium-platinum alloy with molar rate of 1:1 distributed on a VulcanXC-72 carbon compound was adopted to act as the cathode for the electro-catalyst,and the rate of ammonia production was 5.1×10–9g∙s-1∙cm-2at 0.123 VvsRHE,with a decent Faraday efficiency(13.2%)[92].In contrast to solitary Ru&Pt metallic granules,RuPt alloys had synergistic effect towards electrocatalytic synthesis of ammonia,i.e.,N2was trapped on the Ru site,whilst Pt-H supplied hydrogen,following the reaction below (Eqs.(1)-(3)):
Bimetallic/alloy nanostructures could display characteristics relevant to each of these two separate metals.At a more negative cell potential,the velocity of ammonia generation was dramatically reduced as a result of the hydrogen evolution reaction.By shifting the potential from 0.123 to–0.077 V,the ammonia generation rate increased first and then decreases accompanied by a reduction in FE,which predicted that the NRR dominated at relatively low potentials while competes with HER at elevated potentials.The selectivity of NRR was measured at–0.077 V at 30,50 and 70 °C.Electro-chemical ammonia generation rose along with elevated temperature.The by-product hydrazine signal was consistently low and unchanged,and the RuPt/C electrode was found to exhibit significant stability with a current efficiency of 58%even after 45 h of experimentation.A-palladium hydride had reversible hydrogen storage properties and is a prospective NRR electrocatalyst.Nano porous Pd(np-Pd)was obtained through chemical etching of the alloy,followed byin situhydrogen injection to synthesize nano porous palladium hydride np-PdH0.43(Fig.12(a)) [93].The synthesized nano porous catalysts were subjected to electrochemical texts in 0.1 m phosphate buffer solution(PBS)comprising saturated high-purity Ar or N2.At–0.15 V,the ammonia yield of np-PdH0.43was 20.4 μg∙mg-1∙h-1with FE of 43.6%,almost three times that of np-Pd,and such a multiple was also present in the TOF measurements.np-PdH0.43had an ECSA of 0.39 cm2,17 percent greater than that of np-Pd.Isotope labeling experiments revealed that the H in np-PdH0.43was involved in the ammonia formation.The result of calculation showed that the enzymatic NRR pathway was more appropriate for both np-Pd and np-PdH0.43(Fig.12(b)).The formation of *N2H is the rate-limiting step and the ΔGof the*N2H layer of np-PdH0.43was much lower than that of np-Pd(0.65vs.0.91 eV).np-PdH0.43had a hybridized electronic state analogous to that of np-Pd,but the heterodimerization between the Pd and H atoms resulted in the d-band center of np-PdH0.43being closer to the Fermi energy level.Thus,hydrogen infusion could effectively change the d-electron structure of the Pd atom,thus increasing the stability of the *NxHyintermediate as well as improving the activation.CuAg/Ti3C2MXene was synthesized by the graph method using MXene and Ti3C2as carriers to load copper and silver double metallic particles (Fig.12(c)) [94].Experiments were carried out using rotating disk electrode (RDE) and the prepared catalysts were added to ethanol and Nafion solution and then transferred to a glassy carbon electrode in an electrolyte of 0.1 mol∙L–1potassium hydroxide solution.The ammonia yield and FE of the catalysts was 4.12 μmol∙cm-2∙h-1and 9.77% (–0.5 VvsRHE) (Fig.12(d)).The presence of four elements of Cu,Ag,Ti and C and the successful synthesis of CuAg/Ti3C2were demonstrated by various characterization methods.The DFT calculations revealed that N2was more easily adsorbed on the surface of CuAg/Ti3C2(adsorption energy of 3.34 eV) than Ti3C2(adsorption energy of 2.75 eV) (Fig.12(e)).Moreover,the adsorption energy of H on CuAg/Ti3C2(2.35 eV) and Ti3C2(3.33 eV) indicated that the competing HER processes were suppressed.The continuous distribution of DOS and PDOS surrounding the Fermi energy level of the CuAg/Ti3C2indicated that the catalyst is in a highly conductive metal state,which promoted the rapid electron transfer in the electrocatalytic process,and facilitated the achievement of higher electron conductivity.Compared with other control group,CuAg/Ti3C2exhibited a wider conduction band and valence band overlap.Os elements with the same number of electrons in the d-orbitals as Fe were studied by Yanget al.[132] The nitrogen reduction reaction(NRR) of Os1B11N12/C2N was analyzed and the results showed that the optimized structure of Os1B11N12/C2N was designed(Fig.12(f)).The stability of Os1B11N12/C2N was proved by the binding energy calculation,molecular dynamics simulations,charge density difference and partial states density.Rapid electron transfer might occur during N2activation.Intense hybridization of Os-5d with N-2s and bound state generation after N2adsorption suggested that electron transfer from the N2molecule to the Os atomic 5d orbital,while the Os-5d and N-2P exhibited robust hybridization to each other,with the 2π* orbital of N2gaining electrons through Os-5d,forming occupied or non-occupied orbitals near the Fermi energy level,which usefully attenuated the three-bond of N2.The reaction obeys a distal mechanism.It was evident from the Hirschfeld charge analysis that a positive charge was accumulated in the Os atom,leading to electrostatic repulsion with the proton,which inhibited HER.The BN cluster suitably modified the up-d-band edge of Os to obtain the excellent adsorption intensity on the intermediates,thus boosting up the NRR catalytic behavior of Os1B11N12/C2N.
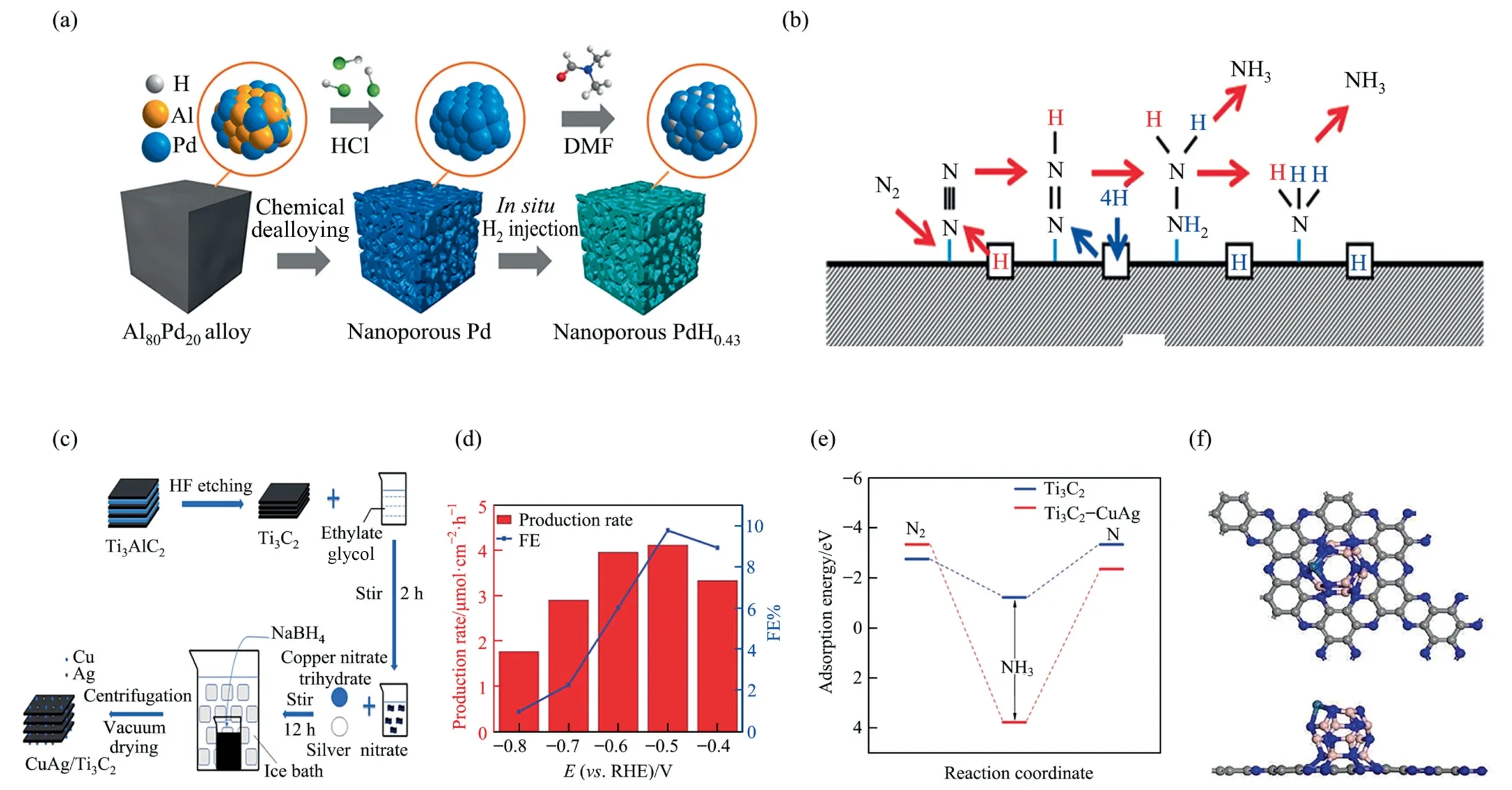
Fig.12. (a)Nano porous palladium hydride synthesis.(b)The Latticed hydrogen reaction route by nitrogen reduction of palladium hydride.Reproduced from Ref.[93]with permission of Wiley Oline Library.(c) Schematic representation of the synthesis of CuAg/Ti3C2.(d)Optimal FE and yield obtained on CuAg/Ti3C2MXene.(e) The adsorption/desorption energies of Ti3C2 and CuAg/Ti3C2 surfaces were computed by DFT.Reproduced from Ref.[94] with permission of Royal Society of Chemistry,copyright 2020.(f)Improved construction of Os1B11N12/C2N.Reproduced from Ref.[132] with permission of Chemistry Europe,copyright 2022.
In general,numerous studies on Au,Rh,Ru and so on are available,whereas a shortage of reports on Ag,Os and Pt was found.
In comparison to Cu,the onset potential ofreduction on Ag is much more negative than that on Cu,demonstrating a higher energy barrier foron the Ag surface.Liuet al.[95] found a strong preference forreduction in the oxide-derived silver(OD-Ag)catalyst,conserving the resultingas a stable product at the electrode.It was calculated that the overall process converted nitrate toin combination withto N2on the Pt catalyst,thus resulting in a FE with up to 98% selectivity and 95%.Amorphous RuO2nanosheets rich in oxygen vacancies were grown on the surface of carbon paper by salt annealing [96].The disordered and vacancy-rich nature caused a shift in the H2atomic centre and d-band centre,which reduced the energy required for ammonia-producing NH2*→NH3*,leading to a FE of 97.46% and an ammonia selectivity of 96.42% by nitrate electroreduction in a 0.5 mol∙L-1NaSO4.Apart from single precious metals,alloying methods are found in recent studies.Wanget al.[97] used a PtRu alloy (Pt75Ru25-C),obtained by chemical reduction of NaHB4,as a catalyst electrocatalytic method for the conversion of nitrate to ammonia.The system exhibited the best nitrate conversion (93%)when pH was 1 and potential was between 0.05 and 0.15 V.The mechanism was attributed to the fact that increasing the amount of Ru in the alloy allows the system to better adsorb nitrate,H2and reaction intermediatesetc.
4.2.Non-noble metals
Noble metals are not the first choice for large-scale industrialization due to its low stocks and high costs.The non-noble metals are plentiful and are considered as prospective materials by virtue of their accessibility to synthesis,cost-effectiveness and even mixed e-ion conductivity properties [133].Therefore,they have been heavily studied.The unusual d orbitals and enriched electron of TM facilitate the weakening of N≡N [134].However,it also benefits the formation of metallic hydrogen bonds,interface engineering,doping and structural defects are required to improve catalyst selectivity [102,135].
Conget al.[98]dissolved Nafion solution,N-doped carbon nanotubes/Fe3C nanoparticles(NCF)obtained by treatment with P123,melamine and iron nitrate in isopropyl alcohol and ultrapure water to make a homogeneous catalyst ink which was attached to nickel foam electrode.Nafion117 membrane was used to separate the cathode and anode with Pt foil as counter electrode and Hg/HgO electrode as reference electrodes (1 mol∙L–1potassium hydroxide electrolyte).The ammonia yield was 15.804 μg∙mg-1∙h-1and FE was up to 2.72%at–0.4 V.High-angle annular dark field (HAADF),SEM,TEM,XRD as well as XPS revealed the morphology of carbon nanotube and attached iron-based nanoparticle,giving the possibility that iron and carbon combine together to form iron carbonation as active sites.The catalytic activity increased with the increase of iron content.However,iron affected the activity by leading to agglomeration of iron nanoparticles.The distinct hysteresis loop in the isotherm of nitrogen adsorption–desorption experiments indicated the coexistence of micro/mesoporous in NCF.A high percentage of doped pyridine nitrogen promoted N2adsorption,which increased hydrophilicity and enhanced electrolyte–electrode interactions.Therefore,it could lower the activated energy of nitrogen reduction and accelerated the electrochemical ammonia synthesis [136].Heet al.[99] prepared ferric tetroxide by electrochemical means (Fig.13(a)).A tripleelectrode h-shaped galvanic cell with a Nafion film was used for protons transportation to foster the electrolytic process: 0.1 mg ferric oxide nanoparticles were sedimented on carbonized paper(CP)as the working electrode(iron sheets and graphite plates were used as the anode and cathode,respectively).Sodium chloride sulfate (0.1 mol∙L–1) solution was applied as the electrophoresis(Fig.13(b)).In the cathode compartment,ammonia gas electrosynthesis was conducted in the vicinity of iron oxide nanoparticles/CP at certain voltage.The outcome showed that Faraday efficiency of 16.9% was completely accomplished at–0.15 Vvs.RHE with an optimum ammonia yield of 12.09 μg-1∙mg-1.And the primary exchange mechanism was inferred by DFT calculations based on the free energy change (Fig.13(c)).
FeSACs were prepared by TM-assisted carbonization on hard template of silica and characterized by the presence of Fe,N and C elements in porous FeSACs.The absence of Fe clusters or nanoparticles implied that Fe atoms were dispersed in an ndoped carbon matrix [100].The FeSACs were immersed on mirror-polished glassy carbon electrodes.With an overall electric current density up to 35.3 mA∙cm-2,the selectivity of ammonia increased to a maximum of 75%at–0.66 V and could be sustained for 2 h,with a yield of 5245 μg∙mg-1∙h-1.Fe single atomic catalyst effectively prevented N2from producing the required N≡N coupling step by lack of adjacent metal sites,thereby enhancing the selectivity of the ammonia product.The main by-product of nitrate reduction by FeSAC was,with Faraday efficiency ofreaching up to 66% and subsequently decreasing to a minimum of 9%accompanied by a rise in Faraday efficiency of NH3,indicating thatmay be an intermediate product that might be further reducible to ammonia.DFT calculations were performed to investigate the reaction mechanism (Fig.13(d)): during the reaction+H++8e–→NH3+H2O,the efficient active site of FeSAC,i.e.,Fe-N4center,optimized electrocatalytic conditions including potassium nitrate,pH electrolyte,and applied potential together imparted the superior electrochemical properties of the material.Unique construction of F-Fe:TiO2was obtained by soaking Fedoped nanoparticles of titanium dioxide obtained by solute-gel process in sodium fluoride solution (Fig.13(e),(f)) [101].Afterwards F-Fe: TiO2was mixed into Nafion solution to fabricate ink loaded on CP.XRD/SEM and other electron microscopy interpretation showed that Fe and F elements were distributed equally throughout the nanoparticles without any aggregation and change in the crystal phase during the reaction.

Fig.13. (a) Specific reactions in the electrochemical process of magnetic ferric tetroxide catalysts.(b) electrochemical device.(c) Free kinetic energy shift of NRR on the Fe3O4(3 1 1) surface.Reproduced from Ref.[99]with permission of Elsevier,copyright 2021.(d) The minimum energy pathway that results in NH3 as the main product.(e)Synthesis schematic of Fe SAC.Reproduced from Ref.[100] with permission of Nature Publishing Group,copyright 2021.(f) Schematic diagram of F-Fe: TiO2 cell.(g) Free energy change graph and reaction cycle diagram of N2 hydrogenation on the surface of F-Fe:TiO2(101)at U=–0.50 V.Reproduced from Ref.[101]with permission of Elsevier,copyright 2022.(h)SEM image of Fe3O4@MoS2.(i)Ammonia production efficiency and FE of Fe3O4@MoS2 at various potentials.(j)Charge density difference of Fe3O4@MoS2 system.(k) Free energy diagram of the electric reduction of N2 at the Fe3O4@MoS2 interface calculated by DFT.Reproduced from Ref.[102] with permission of Elsevier,copyright 2022.
Density flooding theory calculations showed that a defective energy level was formed near the Fermi energy level by introducing Fe atoms,and superficial receptors energy levels might contribute to electron trapping and delivery.The increment in the degree of dissymmetry of the ligand environment to the Fe atom was dependent on the partial replacement of the oxygen atom on the surface by the fluorine element which also intensely appealed to electron cloud,thus decreasing the quantity of oxygen atom bound to the Fe atom and decreasing the work function.After remodeling by F,the d-band center of Fe shifted to higher energy close to the 3-valent Fe,which caused an upward shift of the trap state level position,increasing the bias of the adsorbed N2(*N2)from the trap state to the anti-bonding 1πg*orbital.Thus,F atoms on the interface further customized the local electronic structure of the Fe sites,which could assemble N2molecules [137].The quadrupole splitting of two double states found in the sample from the Mossbauer spectrum foreshadowed the replacement of the Ti4+site by the high-spin state Fe ion.The electrons in the orbitals modulated the spin state of the surface iron ions and thus weakened strongness of the N≡N bonding.The maximum ammonia production velocity was 27.86 μg∙h–1∙mg-1and the highest FE was 27.67%at–0.5 Vvs.RHE,a schematic diagram was presented.The schematic diagram obtained from the calculations is shown in Fig.13(g).Composites of nanospheres of porous ferric tetroxide supported by molybdenum disulfide (Fe3O4@MoS2) were synthesized with formation of a homogeneous mosaic structure (Fig.13(h)) [102].Specifically,ferric tetroxide was nanospheres with a porous structure that offers H+channels.N2could be absorbed and reduced on nanospheres that were mounted on MoS2nanoflowers.At–0.5 VvsRHE.the ammonia yield of Fe3O4@MoS2was 73.24 μg∙mg-1∙h-1with FE of 8.22%,which was an outstanding catalytic efficiency (Fig.13(i)).Excessive high and low MoS2content impacted the catalytic efficiency: if the content was considerable high,(1 0 0) side would not easily engage with the reactants;while low content led to the crowded encirclement of ferric tetroxide,which depressed the adsorption of N2.The composite with a Fe3O4:MoS2ratio of 1:6 was capable of exposing more MoS2active surfaces and had the highest ammonia yield and Faraday efficiency.Using DFT calculations,it was found that there was a 0.83e negative charge transfer from Fe3O4to MoS2,resulting in an increase in the charge density of MoS2and a modulation of the fractional density of states of Fe atoms (Fig.13(j)).According to Fig.13(k),it was observed that the ΔGvalue at the Fe3O4@MoS2interface in the initial hydrogenation process from *N2to *NNH was reduced compared to the reference group,and the comparison of the hydrogenation process from *NNH to *NHNH/*NNH2revealed that the alternate pathway is superior to the distal pathway.The exothermic nature of most steps throughout the process also accelerated the reaction.
The studies listed above were designed with the element Mo,inspired by the MoFe protein catalase,likewise recently researchers have advanced a range of Mo-based NRR catalysts,such as single-atom Mo [103],Mo/nonmetal such as Mo2C/NC [104],Mobased bimetallic groups such as MO/W [105],trimetallic groups such as Ni-Fe@MoS2[106].

Fig.14. (a)Schematic diagram of the synthesis process of Mo/HNG catalyst.(b)3 binding site models and their corresponding differences in different charge densities upon N2 adsorption.Reproduced from Ref.[103]with permission of Wiley Online Library,copyright 2022.(c)Catalytic efficiency of different dopants on Mo-W18O49.Reproduced from Ref.[105]with permission of American Chemical Society,copyright 2020.(d)Synthesis steps of Ni-Fe@MoS2.(e)Mechanism of ammonia production by electrocatalytic Ni-Fe@MoS2.Reproduced from Ref.[106]with permission of Royal Society of Chemistry,copyright 2020.(f)Simple diagram of the structure of the reaction process of Y1/NC and Sc1/NC.(g)Free energy diagrams of NRR of distal pathways on Y1/NC and Sc1/NC.Reproduced from Ref.[107]with permission of American Chemical Society,copyright 2020.(h) Top and side views of Ti-VSW-BN and Ti-VNBN N2 adsorption,blue for N,pink for B,and gray for Ti.Reproduced from Ref.[138] with permission of Elsevier,copyright 2021.
Nitrogen-doped graphene(HNG),containing continuous porous backbone and abounding marginal nitrogenous ligand sites comprising pyridine nitrogen and pyrrole nitrogen,prepared by potassium salt assistance as shown in Fig.14(a),was utilized as a substrate for Mo atom valorization [103].And HNG-750 reacted with MoCl5,thereby announcing the anchorage of Mo species.The device was fabricated using Ag/AgCl and sulfuric acid as reference electrode and working electrolyte.At–0.05 V compared to RHE,the electrode comprised of a synthetic electrocatalyst immobilized on carbon fiber paper could obtain an exceptional FE of 50.2%and an ammonia yield of 3.6 μg∙mg-1∙h-1.The Mo/HNG catalyst revealed the coordination between Mo and nitrogen.Theoretical calculations revealed that the presence of edge-coordinated Mo atoms and vacancies on graphene lowered the overpotential for N2reduction,thus improving the catalytic activity of NRR.Fig.14(b) gives three binding modes: MoN3and MoN3+2Vc with a decision potential step differing from MoN4as the hydrogenation step(*N2+H++e–→*NNH)and with a much lower potential barrier than MoN4.The given acidic electrolyte also facilitated the protonation and desorption process of ammonia.The authors concluded that the prepared catalyst followed the distal pathway on the MoN3site.
N-type doped carbon might be generated in ZIFs scaffold by carbonization,forming molybdenum carbides by reaction with the neighboring Mo species [104].Precursors of carbonized bimetallic Mo/Zn-doped ZIFs were initially ready by a two-step method,followed by high-temperature pyrolysis in N2atmosphere for preparation of N-doped porous carbon(Mo2C/NC).Mo2C/NC possessed a characteristic conventional ZIFs originated carbon substrates,presenting a twelve-sided structure featuring rough perforated surfaces and a potential availability of Mo-based products on the Nrich carbon.Mo2C/NC by virtue of the properties of the zeolitic imidazole framework somewhat overcame the harsh conditions necessary for the two-dimensional Mo2C.The highest ammonia yield of 70.6 μmol∙g-1∙h-1and FE of 12.3% for Mo2C/NC catalyst in 0.1 mol∙L–1sodium sulfate electrolyte corroborated the architecture and galvanic stability of Mo2C as well as carbon frameworks derived from ZIFs.Therefore,embedding molybdenum carbide on n-doped carbon nanostructures is a type of the prodigious tactics for the synthesis of productive ENRR catalysts.W18O49nanowires by anisotropic oriented toward (0 1 0) with enriched surface oxygen vacancies displayed enormous catalytic potential.Mo-doped W18O49nanowires were fabricated over a wide pH range [106].As shown in Fig.14(c),FE of Mo-W18O49was 12.1%,which was considerably twice that of pure W18O49at 0.2 V.An apparent electron transition from Mo to W occurred,which promoted the conversions of N2and the production of ammonia by NRR.It is interesting that the graph also verifies that distinct metallic impurities,including Mn,Ce,Sb,Y,Fe,Sn,and Pd,are useful in boosting the ENRR activity.Zenget al.[106] obtained uniform and highly regular cubic morphology Ni-FePBA nano templates by precipitation route,and synthesized Ni-Fe@MoS2by modeling route(Fig.14(d)).MOFs not only facilitated the growth of MoS2nanosheets with 3-dimensional structural templates,but also draw more N2molecules to settle with their wide surface area.The experimental outcomes demonstrated that the hierarchical structure of the material and the tri-metallic synergy endowed the catalysts with a large quantity of exposed active sites,resulting in superior electrocatalytic performance and supported the possible mechanism of NRR on the NiFe-MoS2NC catalyst (Fig.14(e)).In 0.1 mol∙L–1sodium sulfate electrolyte,Ni-Fe@MoS2exhibited a notable ammonia yield of 128.17 μg∙mg-1∙h-1and a gratifying FE of 11.34% at–0.3 V.
Other transition metals (TMs) such as Sc,Y,and Ti are anticipated to possess remarkable ammonia yields.Y and Sc rare-earth SACs were satisfactorily prepared on carbon carriers [107].Sc and Y atoms,having large atomic radii,were fixed on large-scale carbon imperfectionsviasix isomeric bonds of N and C (Fig.14(f)),unlike the generally investigated M-N-C(M=Fe,Co,Ni)SACs with four coordination active sites.The relatively inferior chemical activity of Y-based and Sc-based nanomaterials initially had an exceptional activity by the regulation of the partial electronic configuration of the atoms by virtue of NC.Y or Sc incorporated into the zeolitic imidazolate framework ZIF-8,converting it into SACS with no crystalline carbon structure and highly mesoporous features.The optimum ammonia efficiency of Y1/NC and Sc1/NC were 23.2 and 20.4 μg∙cm-2∙h-1at a lower potential of–0.1 V,respectively.The difference between Y and Sc is that the individual Y active site was much more reactive than that of Sc.And particular crystal faces of Y2O3together with scandium oxide could not validly stabilize and actuate N2owing to the appropriate free energy changes (Fig.14(g)).The speed-limiting step on Y1/NC was the first protonation step.For Y monoaos,the 3N+3C ligand structure is more advantageous than the 4 N coordination.In overall process of Sc1/NC working as the catalyst,the velocitydetermining step was the final process in the formation as well as release of the second ammonia from the Sc atom.A Cu/Ti3C2complex catalyst presented a viable platform for the purpose of designing effective MXene based NRR electrocatalysts [108].It was found by characterization that CuNPs distributed uniformly throughout the Ti3C2structure.Theoretical calculations indicated that the successive DOS distribution of Ti3C2and Cu/Ti3C2next to the Fermi energy level revealed that the catalyst was in a metallic state having great permeability,which was favorable for the rapid electron transition during electrocatalysis.The Cu/Ti3C2composite expressing wider conduction band and valence bands was deposited on GCE as the operating electrode and the NRR was conducted in 0.1 mol∙L–1potassium hydroxide using RDE at 1600 r∙min-1.The ammonia production rate of the prepared Cu/Ti3C2was 3.04 μmol∙h-1with a FE of 7.31% at–0.5 V.It is speculated that NRR may proceed through a distal incorporation pathway based on the hydrazine free generation property.Ti atom-doped stone-Wales defect type and N-defect type boron nitride (BN) bidimensional materials (Ti-VSW-BN and Ti-VN-BN) have been explored lately [138].The stabilization of both doped structures was higher than BN structure,and the ability to adsorb N2was both enhanced,which benefits ammonia synthesis (Fig.14(h)).The results of characterization revealed that Ti 3d,B 2p and N 2p orbitals had a substantial degree of overlap,creating a new bond between Ti,B and N,which enabled the structures further robust.In a similar way,for TiVN-BN,the Ti,B and N orbitals existed some degree of overlap.In Ti-VSW-BN,electron migration from the Ti atom to the surrounding atomic original structure after doping with Ti atoms resulted in an increase in the polarization of B and N,along with a shortening of the B—N bond length.Three B atoms attached to Ti in Ti-VN-BN lost electrons and therefore accumulated electrons on Ti atoms,and the number of electrons attached to N atoms with B was also smaller than that of the original BN,resulting in a shortening of the average B—N bond becomes longer.In Ti-VSW-BN,enzymatic mechanisms were considered more likely to occur in the NRR reaction,whereas in Ti-VN-BN,the NRR reaction might preferably happenviaan association mechanism.
Zhaoet al.[109]electro-precipitated flower-like open structure polycrystalline copper (FOSP-Cu) on carbon fiber paper,involving an 8-electron process during which an inhomogeneous synergistic polycrystalline Cu surface was formed,thereby fully enhancing the liquid-phase mass transfer process and the synergistic catalytic process.The highest nitrate-to-ammonia yield of 101.4 μmol∙cm-2-∙h-1was obtained for FOSP-Cu in 0.5 mol∙L–1Na2SO4under neutral conditions,with a FE of 93.91%.CuPd-MOF,derived by a two-step homogeneous thermal melting method,was reorganizedin situinto a Cu/Pd/CuOxheterogeneous structure for nitrate ammonia production,accompanied by an ammonia yield of 1510.3 μg∙mg-1-∙h-1and a maximum FE of 84.04% [110].The formed multi-phase heterostructure augmented interfacial polarization by exciting heterogeneous space charge fractionation,which led to electron transfer to Cu,facilitating nitrate adsorption on the Pd surface and reduction at the Cu.MOF might provide limited available space to avoid over-agglomeration of Cu atoms,thus causing uniform supersmall nanoclusters,as in the case of the single-atom Cu-MOF,prepared by Xuet al.[111],showing a FE of 85.5%and formation rate of 66 μmol∙cm-2∙h-1for ammonia.The metal could also be combined with organic matter for example to produce a polypyrrole-Cu 3D network(PPy-Cu-E)with a high ammonia yield and a FE of 91.95%[112].The authors concluded that the process of electrochemical reestablishment resulted in PPy-Cu-E possessing a compact arrangement of Cu nanocrystals on its nanofibers,thus enhancing the active surface area.In addition,non-metallic oxides have been substantially developed as catalysts for ammonia production from nitrates,such as Ni2P (NFP) grownin situon nickel foam with unique superhydrophobic surface,metallic properties,low impedance and abundant surface sites for subsequent activity[113];and heterogeneous structured Bi2S3/MoS2nanoarrays synthesized by anchoring Bi2S3nanowires on MoS2nanosheets with interfacial coupling effects,high specific surface area and exposed active sites [114].
4.3.Non-metallic catalysts and three-component composite catalysts
The noble and the non-noble metals catalyst elucidated above are achieved by doping or other strategies,and the corresponding ENRR capacity are improved.The transformation using various metals or other tactics is mainly aimed at improving the adsorption strength of the metal active site to the N2molecule.However,as long as the metal is available then the electrons in the corresponding d orbitals are inclined to create M-H bonds,consequently resulting in the intensification of HER,which is the main competitive reaction of NRR [11].Metal catalysts are still more or less at risk of metal contamination,and in order to circumvent this risk,eco-friendly,alternative metal-free catalysts with high properties have been investigated [120,139,140].Various types of nonmetallic materials have been exploited in recent decades,especially in the last five years,and demonstrate excellent catalytic properties which can comparable to those of metallic materials.There are vast variety of nonmetals that suitable for electrocatalytic ammonia synthesis including organic polymers,carbonbased catalysts,carbon–nitrogen-binding catalysts,B-based,phosphorus-based,etc.
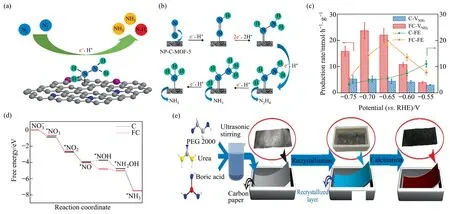
Fig.15. (a) Reaction diagram for simple ammonia production from N and P doped porous carbon.(b) Proposed reaction pathway of NRR on the NP-C-MOF-5 surface.Reproduced from Ref.[115]with permission of American Chemical Society,copyright 2019.(c)Faradaic efficiency and ammonia yield rate of bare C and FC electrocatalysts.(d) Comparison of Gibbs free energy of NO3RR catalyzed by bare C and FC electrocatalysts.Reproduced from Ref.[116] with permission of Elsevier,copyright 2021.(e)Schematic illustration of the formation mechanism of BCN materials.Reproduced from Ref.[99] with permission of Elsevier,copyright 2021.
NP-C-MOF-5 was received by thermal cleavage with dicyandiamide and triphenylphosphine and MOF-5,followed by pickling and washing treatments [115].NP-C-MOF-5 was metal-free and contains rich graphitic carbon.By virtue of the N,P co-doping effect,the defect-rich structure thus generated more dielectric networks,which offered transportation pathways for mass propagation and electron migration (Fig.15(a)).The ammonia output on the NP-C-MOF-5 cathode was 1.08 μg∙mg-1at–0.1 V.Contrary to the other presented catalysts,there was also a notable production of N2H4-H2O during the process with a yield of 5.77 × 10–4μg∙mg-1∙h-1,heralding the generation of N2Hy(1 ≤y≤4) during NRR.Thus,it was concluded that the total ammonia production reaction in NP-C-MOF-5 adhered to the binding mechanism(Fig.15(b)).The fluorine-doped carbon (FC)-synthesized from polytetrafluoroethylene (PTFE) solution after cigarette pyrolysis possessed outstanding NO3RR catalytic performance accompanied by a Faraday efficiency of 20% and an ammonia yield of 23.8 mmol∙g-1∙h-1(at 0.05 mol∙L–1sulfuric acid electrolyte–0.65 Vvs.RHE)(Fig.15(c))[116].The F-dopant,with a low atomic radius and high electronegativity,was allocated in the carbon framework edge with the carbon skeleton to form C—F bonds,disrupting hydrogenation on the active site,which was conducive to the electro-adsorption of nitrate ions through electrostatic interactions.The positively charged carbon atoms after doping homogeneously repelled hydrogen ions and heterogeneously adsorbed nitrate.The consequence of DFT calculations showed that the free energy of the rate determining step in FC electrocatalyst (*NO hydrogenation to *NOH) was lowered in FC electrocatalyst(Fig.15(d)).
Boron carbide (BC) materials was acquired by the procedure of Fig.15(e) [117].The charge redistribution was induced by adjusting the ratio and quantity of boron and nitrogen atoms,which greatly contributed to the chemisorption of the reactants.Significantly polarized B-C σ-bonds resulted in the generation of positive charges on the boron atoms and displays the opposite electronegativity.The boron-rich boron carbonitride(BCN)demonstrated elite NRR capabilities with an ammonia yield of 8.39 μg∙cm-2∙h-1and Faraday efficiency of 9.87% in 0.05 mol∙L–1sodium sulfate under suitable conditions.
S-doped three-dimensional graphene(S-3DG)as a non-metallic galvanic catalyst for NRR at ambient levels was produced [118].The catalytic performance of NRR was markedly improved by 3DG which had superb electronic transfer characteristics and robust physicochemical performances.The 3d-graphene hydrogel,sodium thiosulfate solution and the same pre-chilled mixture(hydrochloric acid,1 mol∙L–1,6 ml)were mixed sequentially to obtain sulfur-graphene gel which dispersed in ethanol–water mixture and Nafion solution after ultrasonication,the evenly distributed ink further was dropped on carbon paper as cathode and electrostatic potential tests were carried out in 0.05 mol∙L–1sulfuric acid with N2saturation.The characterization revealed that the homogeneous doping of sulfur formed carbon–sulfur bonds and maintained the intrinsic interconnected porous network of amorphous 3dgraphene with more folded morphology.The highest yields(38.81 μg∙g-1∙h-1) and FE (7.72%) were observed at–0.6 V.Heteroatomic doping of S atoms with relatively high electronegativity led to a change in the central location of B’s pzorbital (a high energy position away from the Fermi energy level),facilitating the adsorption of N2on the S-CB site[119].The S doping triggered a shift in surface properties from hydrophobic to hydrophilic.The amounts of potassium ions adsorbed and accumulated on the surface of water-friendly electrode rather than on the hydrophobic surface were increased,and the comparatively elevated concentration of potassium ions on the electrode intensely hindered the mobility of protons across the electrode surface from the solution,thus inhibiting side-effect HER.Doping S,which formed double bonds only with C atoms,could induce other defects in the BC3structure which could serve as new active sites.The authors found that CNFs with B and S concentration of 8.09% and 6.23%,respectively,had the highest activity at–0.7 V(FE of 22.4%and ammonia yield of 0.223 μmol∙cm-2∙h-1).Enormously high levels of B could enter the interior of the material to occupy active site of the BC3structure on the surface,leading to a decrease in efficiency.
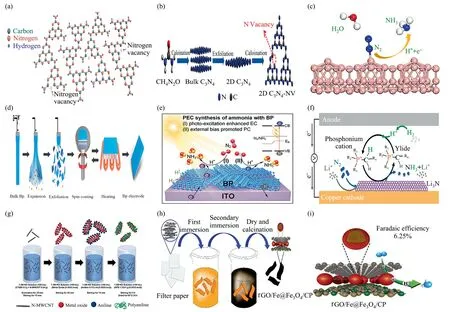
Fig.16. (a) Schematic illustration of NVs engineered polymeric carbon nitride.Reproduced from Ref.[120] with permission of Wiley Online Library,copyright 2018.(b)Schematic illustration of the synthetic pathway for 2D C3N4-NV.Reproduced from Ref.[121]with permission of Elsevier,copyright 2021.(c)Schematic representation of the structure of the ammonia production process from boron nanosheets.Reproduced from Ref.[142] with permission of American Chemical Society,copyright 2019.(d)Schematic illustration of the synthesis of BP NSs and BP electrodes.(e) Sketch of the mechanism of PECNRR enhancement in BP under illumination.Reproduced from Ref.[122]with permission of Wiley Online Library,copyright 2020.(f)Schematic diagram of H2 and N2 sustainable electrical ammonia synthesis.Reproduced from Ref.[124]with permission of Science,copyright 2021.(g)Schematic representation of the synthetic procedure employed for preparation of the HCM electrocatalysts.Reproduced from Ref.[125] with permission of Royal Society of Chemistry,copyright 2019.(h) Flow chart for the preparation of rGO/Fe@Fe3O4/CP.(i) Schematic structure of rGO/Fe@Fe3O4/CP.Reproduced from Ref.[126] with permission of Royal Society of Chemistry,copyright 2019.
In recent years,carbon nitride (CNx) materials have been considered as attractive and hopeful metal-free NRR catalysts due to their outstanding activity,high-temperature stability and costeffectiveness.Liet al.[141]demonstrated a triazine ring defect site in C3N4could be substituted by nitrogen through a range of reactions to generate NH3through isotope labeling experiments.Defects NV to modulate the carbon nitride were explored [120].Polymeric carbon nitrides,prepared from poly-n-ethylbenzene-1,2,4,5-tetracarboxydiimide and porous carbon,were further heated in an argon atmosphere to introduce nitrogen vacancy(NV)defects for improved activity efficiency(Fig.16(a)).The high nitrogen composition and laminar structure of polymeric carbon nitride (PCN)advantageously facilitated the generation of NVs which had the potential effect in regulating p-electron dissociation in PCN covalent systems and might resolve the challenge of achieving strong N2activation.N2could be adsorbed on the PCN in a dual-core end-pair coordination pattern,and electrons on adjoining C atoms were removed to the adsorbed N2,which was the electronic feedback procedure.The incidence of this trend was similar to what usually happens in transition metals.The free energy figure of minimum energy path suggested that the transformation of N2to ammonia favored the mechanism of alternative hydrogenation in the ENRR process.As shown in Fig.16(b),bi-dimensional C3N4-NV with large specific surface area and extensive nitrogen vacancies were prepared from urea by calcination,ultrasonication and lyophilization steps [121].A high volume of pore structure was established during the calcination process,disclosing more active sites,the creation of N vacancies disrupted the long program of C3N4atomic arrangement,which consequently led to the folding phenomenon.At the meantime,the basic structure of the retained triazine unit ring delivers stabilization.DFT calculations revealed that the nitrogen vacancies of C3N4-NV divert electrons to the 2p* orbitals of N2,initiating adsorption and activation of N2while inhibiting HER competition.A distal binding mechanism was suitable.When the potential was–0.3 V,the rate of ammonia generation in 0.1 mol∙L–1hydrochloric solution reached 17.85 μg∙mg-1∙h-1,corresponding to the FE of 10.96%.
A metal-free electrocatalyst (P-atom-doped monolayer C2N)was derived based on the first-nature principle calculations[143].It presents a large cavity structure of N atoms and an enriched lone electron pair.Three classic comparisons of single P-doped C2N inlayers (1P@C2N) and 2P@C2N were first simulated and contrasted.With the various incorporation and spontaneous transition properties of 1P-1,1P-2,and 2P-3,the authors eliminated them from the candidate list leaving only 1P-1 and 2P-1.Bader charge analysis demonstrated the existence of strong interactions between P and N atoms.The sp2hybridization was formed by P atoms in 1P@C2N while sp3hybridization by P atoms in 2P@C2N.NO was preferentially anchored by end-joining method on 1P@C2N,and NO was attached to 2P@C2N at endward and lateral linkages,so the adsorption of NO on 2P@C2N was more favorable for thermodynamic properties.Nitrogen oxidation reduction reaction(NORR)on 2P@C2N was more inclined to the o-distal pathway.All NORR intermediates along the o-distal pathway exhibited a metallic character.The superb conductivity ensured a fast and rapid electron transfer during the reaction and enhanced the catalytic performance.Ammonia synthesized in NORR might be released spontaneously without any additional energy.The weaker adsorption of H atoms on 2P@C2N could well inhibit HER and improved the selectivity of NORR.While 1P@C2N was eliminated for its low selectivity but nice activity of NORR.Boron nanosheets(BNs) as a 2D material have more exposed active sites,moreover have low charge transition resistance and high activity for NRR shown in Fig.16(c).Zhanget al.[142]dropped BNs on carbon writing paper to create BNs/CP which served as a working electrode,BNs at–0.80 V achieved a distinguished FE of 4.04% with high ammonia output (13.22 μg∙mg-1∙h-1) and simultaneously exhibited significant electrochemical durability.Oxidation and deactivation of B atoms of BNs catalyzed NRR more efficaciously compared to the clean BNs,the speed determining step was the deabsorption of the second ammonia gas,as indicated by DFT.The mixture of carbon and phosphorus elements created a stable monolayer carbon phosphide(PCx)that might be a promising electrocatalyst designed by theoretical calculations for PC2,PC3,PC5and PC6monolayer situated on the bumpy hull [144].The PCxmonolayer structure emerged as curved folds offering more surface zone for b-atom adsorption.With a greater carbon content,PCxmonolayers became steadier and all had a smaller internal cohesion energy than BP as well as nano-hole boron nitride.The most propitious adsorption location for an individual B atom on PCxsingle layer(B/PCxSACs)was found by comparing the energies of different adsorption sites.N2was absorbed on the SAC surface where re-distribution of electric charge occurred,electron accretion resulted in self-repulsion of two electronegative N atoms and facilitated N≡N bond activation.For B/PC2SAC,the most beneficial NRR process was the distal pathway.For B/PC3SAC,N2molecule could only be firmly attached through the side-pair conformation and only the enzymatic mechanism was applied.For B/PC5SAC,the alternating pathway was the most thermo-mechanically favorable mechanism.For B/PC6SAC,the NRR procedure as well followed the enzymatic route.The combined analysis concluded that the ranking of the limiting potential was B/PC5>B/PC6>B/PC2>B/PC3.Ab initiomolecular dynamics simulations yielded that B atoms in B/PC2 and B/PC5SACs could occupy the original P-atom positions,significantly disrupting the overall structure,which deprived the possibility to be qualified NRR electrocatalysts.A single B in B/PC6SAC typically could adsorb one N2molecule at a time.Comparison free energy sizes of B with PC6and water molecule yields that B atoms were more favorably anchored to the PC6monomolecular layer rather than being solvated into the aqueous environment.Therefore,B/PC6SAC was considered as the most prodigious candidate system with outstanding reactivity and excellent thermal sustainability at ambient temperature.Black phosphorus (BP),with a plethora of surfaces area and edge positions simultaneously has unfavorable properties for hydrogen absorption,which is exactly what is needed for electrocatalytic ammonia production.At the same time,BP is a straight band gap semiconductor featuring high current-carrying mobility and a wide range of light absorption.BP,which was prepared by assembling exfoliated ultrathin BP nanosheets layer by layer on an indium tin oxide substrate,was used as the experimental electrodes(Fig.16(d))[122].The synthesis approach of BP and rigorous experimental requirements circumvented the nature of BP which was susceptible to water degradation and maintained the stability of BP electrodes.Firstly,the high crystal quality of the ultra-thin BP nanosheets in this work did not readily decompose caused by defects or surface absorbers.Secondly,the cell was well sealed to isolate oxygen,which was a vital ingredient in the BP decomposition process.Light modulated the electronic state of the electrocatalytic active site,and both electrons from outside circuits and optically derived electrons in the conduction band(CB)of BP were involved in the photoelectrochemical (PEC) NRR.The synergistic effects of optical stimulation and peripheral devices made the PECNRR more active than individual photocatalytic (PC) NRR or ENRR,conforming to the mechanism in Fig.16(e).Metal-free,which consisted of black phosphorus quantum dots,conductive polymer(CP)nanofiber diaphragm-loaded BP@CP nanofiber membrane had superior ammonia yields in ammonia activity experiments [123].In addition to the role of the conductive matrix,polyaniline (PANI) was catalytically active without the conductive matrix.BP@CP nanofiber membranes were prepared by electrostatic spinning of polyacrylonitrile/dimethylformamide (PAN/DMF) solutions obtained by adding PANI powder toN,Ndimethylformamide(DMF)treated with internal BP quantum dots.The concentration of BP quantum dots was strictly controlled to avoid severe agglomeration.The weight ratio of PANI/PAN was maintained at 1:4 to ensure the homogeneity of the nanofibers.
The mechanism of Li-mediated NRR is regarded as a viable scheme that relies on the formation of lithium nitride (LiN) at the cathode Li+.A material that can stabilize proton shuttling is required.Bryanet al.[124] invoked a phosphorus-based cation([P6,6,6,14]+) to address this difficulty using platinized titanium as anode,copper as cathode and silver wire as reference electrode.0.2 mol∙L-1LiBF4as electrolyte accompanied by 0.1 mol∙L–1[P6,6,6,14][eFAP] proton shuttle dissolution.Using platinized titanium as anode,copper as cathode and silver wire as reference electrode.0.2 mol∙L-1LiBF4as electrolyte accompanied by 0.1 mol∙L–1[P6,6,6,14[eFAP] proton shuttle dissolution.In 20 h experiments,the authors found that when the voltage was kept at–0.75 Vvs.Li0/+,FE of the reaction was 69%±1%.This debasement process of the cation would produce ammonia and the ylide structure with LiN.Ylide structure was strongly basic and easily reproduces the original phosphine cation with the H+produced in the anodic oxidation reaction,as shown in Fig.16(f).
Catalysts compounded with three types of materials have been equally examined for their peculiar structures.Although the structure contains a small number of metallic elements,they are still classified here according to the properties of their main bodies.The Fe3O4/PANI/N-doped-multi-walled carbon nanotube (NMWCNT) and RuO2/PANI/N-MWCNT,prepared by the method shown in Fig.16(g),were the first two hybrid composites for electro-catalytic ammonia production in moderate circumstances[125].The oxide precursors (triton tetroxide and RuO2) were homogeneously distributed on the PANI/N-MWCNTs in the form of particles without any free aggregates.The voltage values of Fe3O4/PANI/N-MWCNT or RuO2/PANI/N-MWCNT at which the FE and ammonia synthesis rate reached the maxima value were not coincident at 25 °C.Fe@Fe3O4/CP with sandwich-like could reduce graphene oxide with eggshell structure by impregnationcalcination strategy successfully (Fig.16(h)),which could be directly used as a self-supporting electrode,high electrocatalytic activity (1.3 × 10–10mol∙cm-2∙s-1) and superb alternative (FE value is 6.25%) were obtained [126].rGO/Fe@Fe3O4/CP exhibited a characteristic three-layer structure(Fig.16(i)):Ultrathin reduced graphene oxide nanosheets that suppressed the agglomeration of particles during calcination uniformly wrap the inner CPs,and the Fe particles present between them were unevenly anchored on the surface of reduced graphene oxide nanosheets.The external yolk-shell nanostructure realized involvement of Fe@Fe3O4nanoparticles in the reaction and isolated the internal Fe nanoparticles from the intermediate avoiding the poisoning of Fe sites and corrosion of the electrolyte.
5.Photocatalytic Syntheses of Ammonia
When we mention photochemical synthesis of ammonia,The first thing that comes to mind is the nitrogen fixation from nature by plants through photosynthesis catalyzed by enzymes,but this procedure is not compatible with the requirements of contemporary technology for ammonia synthesis owing to its slowness.The natural light nitrogen fixation enzyme,molybdenum nitrogen fixation enzyme,is a multiple emergence from a crowd of enzymes and consists of two individual proteins [145,146].The smaller of the two,referred to as ferritin (Fe protein),comprises one metal center (a single [4Fe-4S]2+/+cluster);the larger one,entitled molybdenum-iron protein (MoFe protein),contains 2 metal centers: the p-cluster (each tetramer having two),each containing 8 pairs of bridging connected iron atoms with S2–atoms ([4Fe-4S]cluster);the Mo:Fe:S atom ratio of the M-cluster(FeMo cofactors,each tetramer having two),is 1:7:9 (Fig.17(a),(b)).It is generally believed that in nitrogen fixation,the orientation of electron flow is from the original ferritin protein to the P-group,then to the M-cluster and last to the substrate.Each electron shift involves the hydrolysis of at least two ATP molecules [145,148].Ironbased proteins hydrate adenosine triphosphate(ATP)to adenosine diphosphate (ADP),and the generated electrons are available to MoFe based proteins,in which N2is converted to NH3[13].This is commonly described as follows (Eq.(4)) [148]:
It is evident that nitrogen fixing enzyme metal clusters are pivotal in electron transfer and substrate reduction.From the energy band perspective,in the photocatalytic synthesis of NH3,the valence band (VB) holes oxidize water and the conduction band(CB) electrons actuate the N2reduction reaction,which trigger the inspiration to mimic metal clusters for photochemical reactions.The crucial point is the need to discover a semiconductor photocatalyst that can effectively absorb the solar energy efficiently and squeeze the energy adequately to generate electrons for the reduction of N2to NH3under ambient conditions[81].Photosynthesis of ammonia can be classified into photochemical system type and photoelectrochemical model (Fig.17(c),(d))[81,147,148].In the former,nanoparticles are dispersed in N2-saturated aqueous solution to form a suspension as a photocatalyst,where electron is excited with the energy of light wider than the forbidden band width excites,leading to the occurrence of redox reactions at diverse locations on the particles.On the other photochemical model,as in the case of electrochemical ammonia production,the oxidation–reduction reaction proceeds separately.Electrons and holes are induced at the photovoltaic electrode to foster charge separation.Nitrogen fixation is performed at the surface of the photocathode.The process of nitrogen fixation on either photochemical or photoelectrochemical model may be viewed as three-part process: Firstly,light irradiated on the surface of the material,causing the excitation of photon with increased energy,the generation of vacancies and electrons.Secondly,carrier charges are not compounded and migration occurs.Thirdly,the carriers mobilize to the active sites on the catalyst surface and reduce the N2molecules adsorbed on the active sites to ammonia[81,149,150].It is noteworthy that a rather negative electrode potential is still necessary for NRR to occur,which is arduous to achieve with the conduction band of common photocatalysts (e.g.,TiO2) leading to difficulties in solar-driven nitrogen fixation.Therefore,the hunt for potentially suitable photocatalysts has become a coveted goal of researchers of generations.Table 5,Table 6,Table 7 depict the properties of the recent ammonia photocatalysts outlined in this paper.
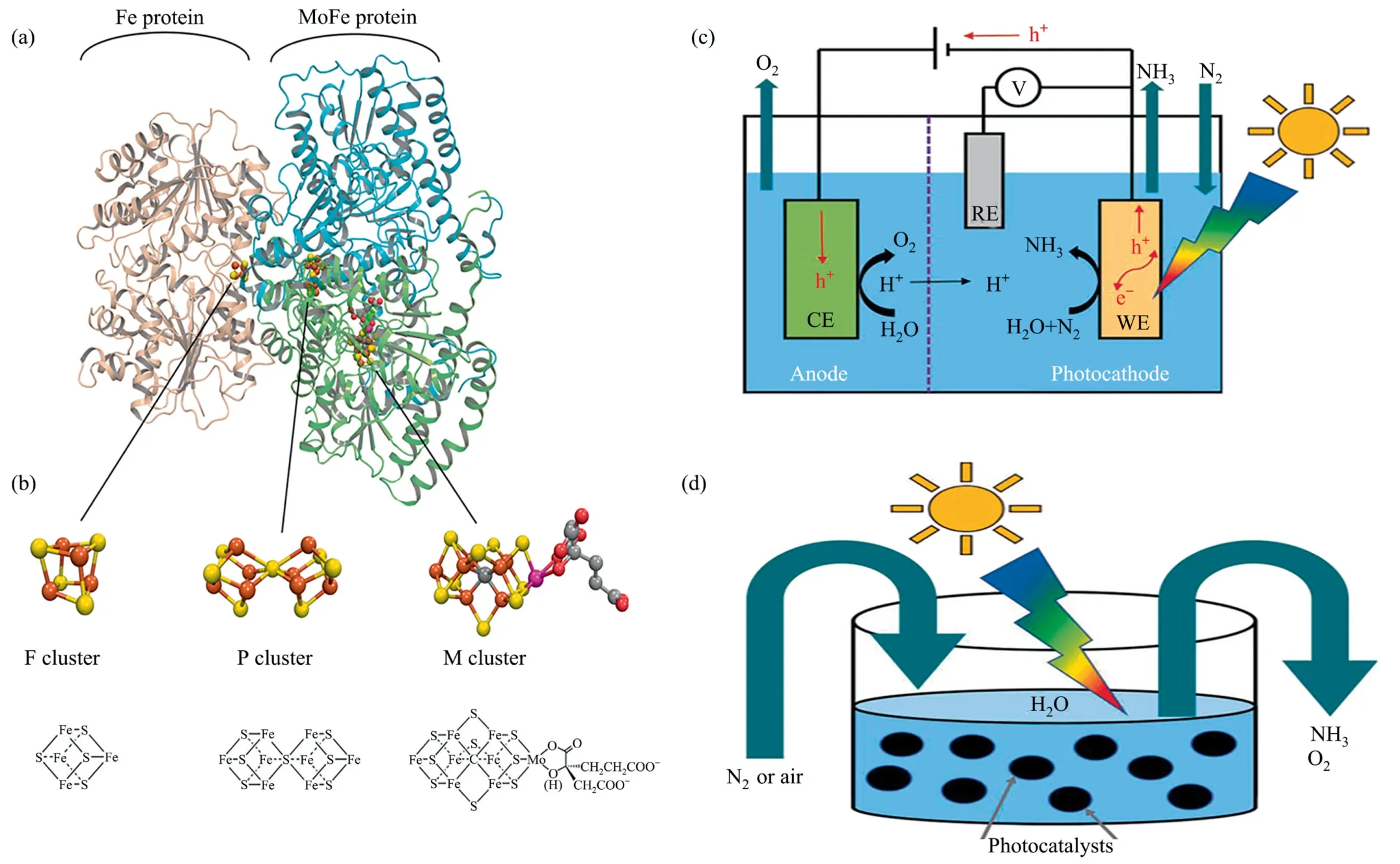
Fig.17. (a)MoFe protein complex and Fe protein homodimer in molybdenum nitrogenase.(b)Fe protein(F cluster),MoFe protein(M cluster)and a Fe protein homodimer(P cluster).Reproduced from Ref.[13]with permission of Elsevier,copyright 2019.(c)Device model of a photocatalytic reactor.(d)mock-up of a photocatalytic reactor for the photochemical production of ammonia.Reproduced from Ref.[147] with permission of Nature Publishing Group,copyright 2013.
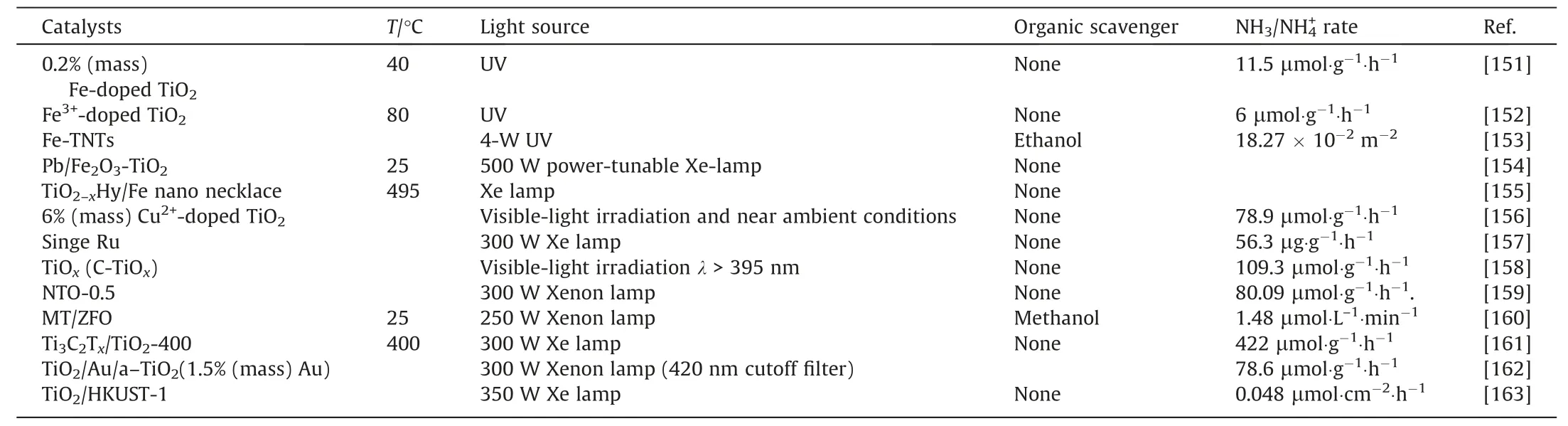
Table5 Summary of the TiO2-based catalyst performance mentioned in this paper

Table6 Summary of the Bi-based catalyst performance mentioned in this paper

Table7 Summary of the other emerging catalysts performance mentioned in this paper
5.1.TiO2 base materials
The use of TiO2in photocatalysis traces back to the 1940s,when Dhar and Pant conducted experiments on sterile soil and concluded that the lynchpin for the fixed conversion of N2to NH3was the presence of light and metal oxides (e.g.,TiO2,ZnO and Fe2O3) [185].The development seemed to be stagnated until the 1970 s,when Schrauzer and Guth [151] first systematically investigated the photocatalytic synthesis of ammonia.an ironcontaining titanium dioxide catalyst prepared by heating the mixture of TiO2and iron(III) sulfate,was subjected to ultraviolet (UV)light and was found to reduce the adsorbed N2to produce NH3in small volumes of hydrazine.And it was observed that the optimum efficiency of ammonia production occurred at 0.2% (mass) of iron doping.This phenomenon could be explained by the increase of rutile structure in TiO2as a consequence of the increase of iron content,which dominated the growth of efficiency.And the loss of larger particle size and specific surface area changed into the prevailing factor to reduce the efficiency,when the iron content exceeded 0.2%(mass)[186].The reorganization of photogenerated electron-hole pairs could be inhibited by the inclusion of Fe3+as an impurity,thus improving the photocatalytic activity.In 1988,Bourgeoiset al.[187]realized that pure rutile or anatase structured TiO2powders didn’t generate ammonia when subjected to UV irradiation in the presence of moist nitrogen,and the anatase-to-rutile phase transition was determined by the nature of the precursors used.In order to undergo the photochemical reaction,the sample first had to be annealed at high temperature,which caused defective states in the band gap and hydroxyl groups on the surface of the semiconductor.Cooling from higher pretreatment temperatures (>1000 °C) was essential to induce surface defects in the TiO2material and small quantities of iron contamination (<1%)were essential to improve ammonia production.Oxygen vacancies on or near the surface of nanoparticles could enhanced their adsorption to water and formation of surface hydroxyl groups,thus improving the photocatalytic activity.However,when the photocatalyst was over-doped with Fe3+,the temporary photogenerated electron or hole trapping sites would disappear and Fe3+gradually acts as the complex center [152].The complexation of trapped electrons and holes increases,thus decreasing the photocatalytic activity.Augugliaroet al.[188] used an impregnation-calcination method to synthesize γ-Al2O3-loaded Fe2O3/TiO2hybrids with good ammonia productivity.
Zhaoet al.[153]highly modified TiO2with Fe3+to create surface oxygen vacancies which exhibited efficient N2photo immobilization (Fig.18(a)).Titanate nanotubes in the titanate hydrogen(H2Ti3O7) phase were prepared by ion exchange method.Irondoped titania nanoparticles with widely available high exposure(1 0 1) surface were immaculately obtained by a dual-step hydrothermal approach using TNTs as precursors for iron adsorption.Ammonia concentration and quantum yield production increased with ethanol concentration (Fig.18(b)).And a possible electron transfer mechanism on Fe doped TiO2nanoparticles was proposed (Fig.18(c)).Thanks to the promising effect of ethanol in removing ∙OH,ammonia could not be oxidized to its higher oxidation state during the reaction when it was a scavenger,causing no formation of nitrite or nitrate.Lashgariet al.[154] proposed the synthesis of some homogeneous nanoparticles based on Fe2O3and TiO2by a simple precipitation/calcination method,after that dissolved palladium acetate in ethanoic acid.The previously synthesized binary photocatalyst was scattered in aforementioned solution by irradiation of xenon light source,after the photo deposition of palladium,tricatalysts (Pd/Fe2O3-TiO2) was obtained.The transcendence of Pd/Fe2O3-TiO2could be understood as follows:the catalyst formed a metal/semiconductor Schottky barrier resulting in a better charge separation which facilitated the delivery of light-generated electrons to the metal sink,while the excellent H-atoms storage capacity of palladium along with the larger apparent area of the photocatalyst enabled more nitrogen molecules to be trapped (Fig.18(d)).
Ammonia production from two-temperature zone centers based on TiO2materials has also been explored.Maoet al.[155]used the centralized diffusion of the solar flux of nanostructured metallic Fe (nano-chain microstructure) with localized surface plasmon resonance effect to generate heat in a small volume as a N2dissociation center.A well-aligned center with low thermal conductivity (TiO2–xHy) was picked to form a ‘‘low temperature”zone when it was coupled.TiO2–xHynanoparticles prepared by reduction of NaBH4and hydrogenation of TiO2produce TiO2–xHy/Fe by reduction of FeCl3(Fig.18(e),(f)).EPR measured TiO2–xHycontaining oxygen vacancies.By virtue of weak physical adhesion,TiO2–xHynanoparticles were robustly bound to Fe nano strands through lattice layer-mediated interactions of amorphous transition iron oxide (Fe2O3–d),thus achieving homogeneous anchoring on Fe nano strands.Solar-catalyzed ammonia synthesis was performed on TiO2–xHy/Fe nanocomposites,Fe nanocomposites,titanium dioxide/Fe and conventional Haber iron catalysts.Precise modulation of O vacancy concentration in titanium dioxide nanosheets was achieved by simple doping of TiO2nanosheets with Cu2+(Fig.18(g)).These Cu-doped ultra-thin TiO2nanosheets displayed outstanding properties for photocatalysis.The optimum copper loading was 6%(mass),and the catalyst extended the activity towards light to 700 nm achieving a high ammonia productivity of 78.9 μmol∙g-1∙h-1under full solar irradiation(Fig.18(h)) [156].

Fig.18. (a)PL spectra of undoped TiO2 and Fe doped TiO2:PL peaks at 408 and 440 nm are caused by surface oxygen vacancies and defects due to Fe3+doping.(b)Influence of ethanol concentration on ammonia production in Fe3+-doped titanium dioxide(S100)under neutral conditions at room temperature.(c)From N2 to ammonia processes on the surface of titanium dioxide(1 0 1).Reproduced from Ref.[153]with permission of Elsevier,copyright 2014.(d)Probable reaction pathways for photo-fixation of N2 molecules at the semi-conductor/solution interface in the vicinity of sacrificial agents.Reproduced from Ref.[154]with permission of Elsevier,copyright 2017.ⓒIllustration of TiO2–xHy nanoparticles bonded to the Fe-nano clad surface to form TiO2–xHy/Fe hybrids for photothermal ammonia synthesis.(f) STEM image of TiO2–xHy/Fe.Reproduced from Ref.[155]with permission of Elsevier,copyright 2019.(g)2D structural model of titanium dioxide nanosheets with Vo.(h)Diagram comparing the ammonia activity of TiO2 doped with different concentrations of Cu.Reproduced from Ref.[156] with permission of Wiley Online Library,copyright 2019.
Ruthenium is favored for its low N2reduction overpotential compared to Fe.The synthesis of oxygen vacancy-rich singleatom Ru-modified TiO2nanosheets has been previously reported[157].For primitive TiO2without oxygen vacancies,Ru was mostly contained in clusters and small nanoparticles,and relatively large Ru nanoparticles were observed at higher Ru loading (2% (mass))(Fig.19(a)).Ru was inlaid in the lattice in Ru-doped TiO2nanosheets with oxygen vacancies(TR-NS).The single-atom metal distributed on the carrier homogeneously with low metal-atom coordination environment and the highest metal utilization efficiency.The valence state of Ru was close to 3 and there was no dispersal between Ru and Ru,the concentration of O vacancies dropped compared to TR-NS because of the occupation of individual Ru atoms in O vacancies.The shift from 2.34 to 2.48 eV at the top of the VB of TR-NS with 1.0% (mass) Ru (TR-1.0) was presumably attributed to the interaction between TiO2and Ru(Fig.19(b)).The authors concluded that Ru species might displace water molecules bound to oxygen vacancies,thus interfering with the transport of photoelectrons from TiO2to H+and inhibiting HER.In addition,the vacant d orbitals of Ru could receive light electrons to minimize the complexation of electron-hole pairs.A single Ru site dramatically promoted the photoreduction of N2to ammonia.The photocatalytic activity of monoatomic Ru-loaded TiO2nanoflakes for N2immobilization was detected by the light source of a 300Xe lamp.Loading 1%(mass)of Ru on TiO2nanosheets remarkably improved the photocatalytic N2reduction.The photo-catalytic N2reduction capacity of auxiliary catalysts in four precious metals(Ru,Rh,Pt,Pd)had been examined a long time ago and the authors determined that the reduction ability was linked to the strength of the mental-hydrogen bond following the principle Ru >Rh >Pd >Pt (Fig.19(c)) [189].The porous C-doped TiOxnanosheets prepared by H2O2-assisted thermal oxidative etching(H2O2-TOE) strategy which used layered Ti3SiC2as a structureguided template and doped with sources of C and Ti had come into the research limelight (Fig.19(d)) [158].The influence of carbon doping in TiO2on activity was summarized into two aspects:accelerating the cleavage of N≡N and narrowing the band gap of TiO2.The considerable content of Ti3+sites generated in the process of C doping into TiO2by bottom-up substitution was used as the primary contribution to increase the N2photoreduction activity of C-TiOx.The bottom-up strategy allowed for excellent control of the solubility of the substituted dopant in the bulk and was repeatable.The porous nanosheet morphology increased the number of active catalytic sites as a result of its considerable BET,and the presence of Ru/RuO2nano catalysts provided a synergistic effect on the C-TiOxand enhanced its charge separation and transfer capability.All the C-TiOxsamples produced exhibited high photocatalytic ammonia production rates,C4-TiOx(109.3 μmol∙g-1∙h-1)far exceeded the other carbon content samples,where the Ti3+/Ti4+ratio was 72.1%,which was far ahead of all TiO2photocatalysts.The combination of photo-catalysis and thermal catalysis between the Ni and TiO2materials enabled the production of ammonia under uncritical conditions (Fig.19(e)) [190].The reaction was characterized by the formation of NiO/NiO2and the simultaneous change of Ti4+to Ti3+generating oxygen vacancies,which influenced the reaction process of N2molecules and the capture of photoelectrons on the TiO2,while the Ni atoms are capable of dissociating H2molecules and retaining the vacancies.The Ni/TiO2catalyst was analyzed by XRD and the signals of TiO2peaks in the type of anatase and rutile with Ni were detected,and the average dimensions of anatase and rutile were calculated to be not much difference (over 30 nm),and the average dimension of Ni nano-particles was 10 nm.The UV-spectra of Ni/TiO2demonstrated that the Ni/TiO2catalyst contributed more to the absorbance of visible and near-infrared radiation.Photoelectrons trapped in vacancies on Ni/TiO2activate N2molecules,H atoms formed by chemisorption and dissociation of Ni nanoparticles were migrated to the Ni-TiO2boundary,where they hydrogenated with activated N2to produce intermediates further hydrogenated to ammonia.N-doped TiO2hollow microspheres (NTO-0.5) containing vacant sites for oxygen were synthesized by water heat method adopting phenolic resin microspheres as a matrix,Ti(SO4)2and resorcinol as resource (Fig.19(f)) [159].The ammonia yield of the resulting NTO-0.5 was 80.09 μmol∙g-1∙h-1.The explanation for this was as follows:(1)the hollow structure enhanced the light utilization;(2)the n doping narrowed the band gap(3.18–2.83 eV)enhancing light uptake and usability;(3) the oxygen vacancy of NTO-0.5 induced a positive surface charge and promoted the adsorption of N2;(4)more photogenerated electrons were electrostatically attracted to the surface by the positive charge,which were then transferred to the superficial layer and participated in the light-catalyzed reaction (Fig.19(g)).

Fig.19. (a)SEM image of TR-1.0.(b) Schematic diagram of the energy band change and ammonia production process in titanium dioxide nanosheets decorated with single ruthenium atoms.Reproduced from Ref.[157] with permission of American Chemical Society,copyright 2019.(c) Relationship between the strength of the M-H bond and the yield of ammonia(1 kcal=4.184 kJ).Reproduced from Ref.[189]with permission of Elsevier,copyright 1996.(d)Procedure for the preparation of C-TiOx.(e)Photothermal nitrogen hydrogenation route over Ni/TiO2 catalysts.Ref.[190] with permission of American Chemical Society,copyright 2021.(f) Synthesis process diagram for NTO-0.5 hollow microspheres.(g) Potential Nitrogen Photo-fixation Schemes for NTO-0.5.Reproduced from Ref.[159] with permission of Elsevier,copyright 2022.
Ronget al.[160] reported a z-type titania/ZnFe2O4heterojunction photocatalyst with greater photocatalytic activity than either titania or ZnFe2O4alone.ZnFe2O4(ZFO)loaded with modified titanium dioxide (MT) was obtained using a solvothermal calcination method to form a direct solid z-format system.Under ambient temperature and pressure and in methanol,the enhanced photocatalytic performance of the MT/ZFO with ammonia production speed of near to 1.48 μmol∙L–1∙min-1was attributed to the negative reduction potential of the z-type charge transfer model in the ZnFe2O4conduction band caused by the photoinduced VB photocatalytic decomposition of methanol.The charge separation was enhanced by double charge transfer mechanisms (Fig.20(a)): In model 1,photoinduced electricity in the CB of ZnFe2O4was transferred to the CB of titanium dioxide,while photoinduced holes in the VB of titanium dioxide were moved to the VB of ZnFe2O4.In model 2,the solid–solid contact interface between titanium dioxide and ZnFe2O4was the center of the photogenerated electronhole complex.The photo-induced bores in the VB of titanium dioxide were interacted with water to form H+which reacted with N2in the CB of ZnFe2O4and the photo-induced electrons to form ammonia (Fig.20(b)).Recently plasmonic and TiO2heterostructures have been similarly investigated for Ti3C2TxMXene,a 2D TM carbide and nitride [161].In specific,titanium carbide has properties of superb electrical conductivity and tunable optical characteristics.Houet al.[161] combined TiO2and Ti3C2TxMXene and utilize the features of Ti3C2Txin visible and near-infrared photocatalysis,which addressed the major obstacles of its inferior vehicle splitting performance and poor active site.Ti3AlC2powders were subjected to selective etching by 40% hydrofluoric acid at indoor condition of the Al layer for 72 h,then ultrasonicated in a 400 °C muffle furnace to obtain the hybrid structures.Photocatalytic irradiation of the resulting Ti3C2Tx-25 with a xenon lamp under full spectrum irradiation at four temperatures revealed that the NH3yield of Ti3C2Tx/TiO2-400 was 422 μmol∙g-1∙h-1at 400°C.More substantially,under NIR light,the ammonia yield of Ti4C2Tx/TiO2-400 at 740 nm monochromatic light alone reached 82 μmol∙g–1∙h-1.The plasma Ti3C2TxMxene phase in Ti3C2Tx/TiO2-400 realized the capture of NIR light to form thermal electrons concomitant with vacant space for oxygen in the TiO2phase of Ti3C2Tx/TiO2-400 as an effective center for potent attachment and activation of N2.Ultrathin titania nano-sheets decorated with gold nanocrystals permitted efficient photoreduction of N2to ammonia under visible illumination,gold nanoparticles uniformly spread over the surface of the titania nano-sheets [191].The selfsynthesized photocatalyst markedly improved the portable accessibility of ammonia synthesisviaa ‘tandem’ pathway in which the TiO2nanosheets on oxygen vacancies could activate N2molecules,followed by the reduction of ammoniaviaE-generated by plasma stimulation of Au nano-crystals (Fig.20(c)).Liet al.[162]inserted surface oxygen vacancies into plasma-enhanced rutile TiO2/Au photoelectrodes to form a slim amorphous TiO2in the external layer (Fig.20(d)).This particular arrangement (TiO2/Au/a-TiO2) promoted the conversion of N2to ammonia at a speed(13.4 μg∙cm-2∙h-1),indicating co-operative interaction between photo absorption augmented by plasma and N2sorption/activation triggered by surface oxygen vacancies.Wanget al.[163] prepared by the steps shown in Fig.20(e) a thin-film photocatalyst that might be used in a gas–liquid-solid three-phase reaction scheme based on HKUST-1 thin film.It has advanced nitrogen adsorption capability(Fig.20(f)),supported by TiO2nanoparticles with photocatalytic nitrogen sequestration performance.The efficiency of ammonia synthesis was up to 0.048 μmol∙cm-2∙h-1,which was 2.8 folds higher than that of the TiO2/CM (copper mesh) catalyst under the same conditions.The membrane catalyst was free of nitrogen and would not disturb the ammonia synthesis reaction.
In summary,there has been considerable research on TiO2-based photocatalysts to improve the efficiency of photocatalytic ammonia synthesis by means of doping,compounding and plasmaisation.
5.2.Bismuth-based materials
Bi-based substances are also extensively investigated for their advantage of changeable energy band gap,non-toxic and excellent degradation ability and high-availability of solar energy,including bismuth metal,bismuth oxide,bismuth oxyhalides,bismuth molybdate,etc.[192].
Bi is a less reactive hydrogen evolution reaction(HER)material that significantly promotes nitrogen fixation by blocking competing HER.Bi/InVO4photocatalyst with a 5.2-fold increase in photocatalytic ammonia yield compared to pure InVO4was reported by Wanget al.(Fig.21(a),(b)) [164].Bi2O3has been used as a photocatalyst already for a considerable long time and is incomplete fluorite stacking structure which originally has only 6 cations filling the positions of 8 anions in the cell hence there are 2 oxygen dot positions without ions on them.Therefore,these empty sites can catch N2by chemisorption and then be activated by electrons generated by light energy.The availability of 6 s2solitary pair electrons in δ-Bi2O3,the strong deformation and polarization of Bi—O bond are favorable to electron migration,which makes the Bi2O3-based photocatalyst have excellent photocatalytic N2reduction performance [193–195].Ag-modified Bi2O3photocatalysts prepared by photoreduction method had multi-layer structures,appropriate absorbing margins and various exposed surface sites,and had the capability of photocatalytic nitrogen fixation on Bi2O3at room temperature and pressure[165].Bi2O3was produced by hydrothermal method employing Bi(NO3)3∙5H2O,ethylene glycol,and tert-butanol as raw material,and different concentrations of AgNO3was dropped to reduce different Ag-Bi2O3particles,which were subjected to 400 W xenon lamp (Fig.21(c)).XRD and SEM confirmed that the lattice parameters shrank along with the growth amount of silver doping,about nanoparticles with the size of roughly 20 um comprised multilayer poly group.Although the crystal network was well defined and homogeneous,the surface roughness turned into small particles and yet each layer became increasingly thin.XPS with EDS indicated the presence of Bi3+and the absence of oxidation status of additional Bi compounds.The productivity of ammonium increased linearly with increasing reaction time,where 0.2% Ag-δ-Bi2O3had the best photocatalytic ammonia production capacity reaching about 5.1 μmol∙L–1,which was about 7 folds of pure δ-Bi2O3.Noticeable traps were grown in the ultra-thin Ag-δ-Bi2O3sheet because the surface plasmon resonance of Ag productively widened the energy band of the photocatalytic catalyst δ-Bi2O3.In irradiation and ultra-fast transition conditions,it was easy to excite electrons and holes and obstacle to expeditious complexation of photo-induced electron-hole pairs to obtain high photoconversion efficiency.
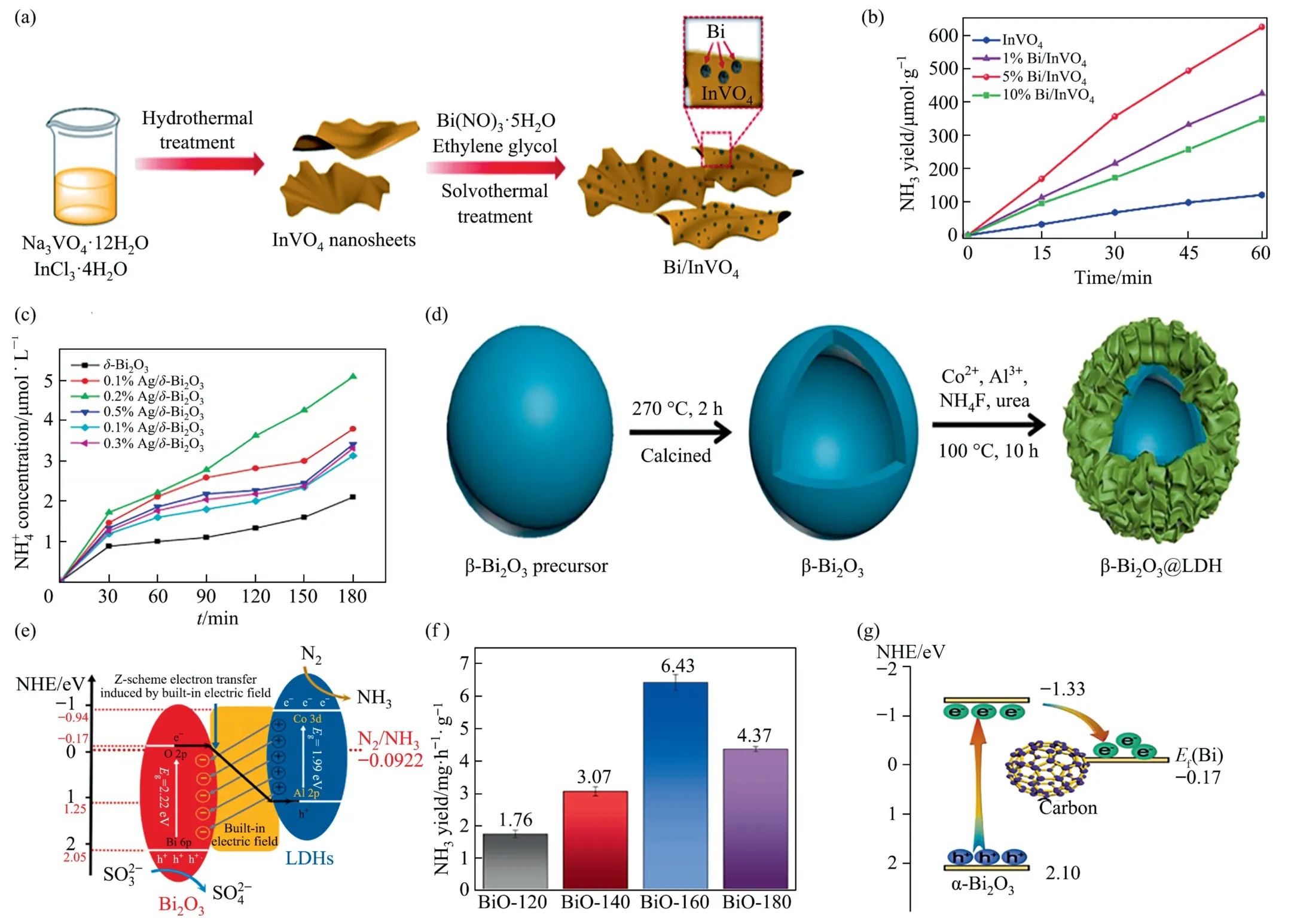
Fig.21. (a)Sketch of the production process of Bi/InVO4 nanosheets.(b)Yields of ammonia from InVO4 and Bi/InVO4 by UV–Vis radiation.Reproduced from Ref.[164]with permission of Royal Society of Chemistry,copyright 2020.(c) Comparison of ammonia yields from Bi2O3 loaded with different amounts of Ag.Reproduced from Ref.[165]with permission of Elsevier,copyright 2019.(d) Schematic diagram of the synthesis process route of BO@CA.(e) Electron conversion mechanism of BO@CAz based hierarchical junction photocatalysts.(f) The ammonia yields of BiO prepared at different temperatures.Reproduced from Ref.[166] with permission of Elsevier,copyright 2021.(g) Energy band structure and possible separation pathways of photogenerated carriers for enhanced photocatalytic activity.Reproduced from Ref.[167] with permission of Elsevier,copyright 2022.
The electrons on the CB of the Z-format heterojunction semiconductor B can reorganize with the holes on the VB of the semiconductor A with the outcome that the photosynthetic electrons are primarily concentrated in the CB of the semi-conductor A and the holes are mostly accommodated in the VB of the semiconductor B,which is the particular electron exchange path to accomplish the effective separation of the spatial photoelectric carriers [166,196,197].Xiaet al.[166] generated a new three dimensional hollow z-type hierarchical junction photocatalyst by Bi2O3prepared by hydrothermal method and co-layered CoAl-LDH prepared by co-precipitation method(Fig.21(d)).The conductive band of LDHs possessed powerful reduction potential for the NRR,which had low carrier transport rate and high carrier reorganization greatly favoring the initiation reaction of N2.The efficiency of ammonia synthesis could reach 48.7 μmol∙L–1∙h-1.The order of ammonia evolution for all samples was BO@CA-1 >BO@CA-2 >B O@CA-0.5>LDH>Bi2O3and the rate of NH4+production was about 147.6 times higher than that of hydrazine.BO@CA had good structural stability and photocatalytic properties.The morphology remained unchanged after the experiment labeling experiments by dimethylformamide (DMF) and acetonitrile instead of water and nitrogen 15 showed that the N and H in ammonia examined in this report were from optical nitrogen fixation.The electrical gap between Bi2O3and LDHs was created by the charge realignment of the heterojunction between LDHs and Bi2O3,which contributed to the formation of a build-up electric field that triggered the migration of photogenerated electrons in the opposite direction.The migration of light-generated electrons from O 2p orbitals to Al 2p orbitals was concomitant with the outcome that photogenerated electrons enriched principally on the CB of LDHs could be effectively trapped in N2to generate ammonium,while cavities enriched mainly on the VB of Bi2O3(consisting of 2p orbitals of Bi elements)could be rapidly arrested by a scavenger sodium sulphatic (Fig.21(e)).It might be deduced that the photogenerated electrons accumulated on the CB of LDHs(Co 3d orbital),indicating that the element Co was the reactive position for the optical nitrogen fixation,which resulted in an enhanced photogenerated carrier lifetime and transposition of photo-activated electrons on the surface.BiO-Bi materials were obtained from Bi(NO3)3∙5H2O and ethylene glycol using a simple one-pot method at variable temperatures,where the sample BiO-Xwas obtained(Xrepresents Celsius temperature)[167].The ammonia concentration was progressively increased with time under simulated solar irradiation with sodium nitrate as the nitrogen source.Among the samples,BiO generated at 160°C(BiO-160)had the most photocatalytic activity with an ammonia yield of 6.43 mg∙g-1∙h-1(Fig.22(f)) and reduced 98.72% of,moreover,the main product was ammonia (95.00%) rather than the hazardous nitrite.As the preparation temperature increasing,the growth of crystals caused a decrease in specific surface area.The lack of oxygen vacancy signal in BiO-160 indicated that oxygen vacancy was not instrumental in the efficient reduction of.At 160°C the glycol would be decomposed in α-Bi2O3leaving carbon elements behind,which improved the electrical conductivity of the composite.In addition,the alcohol hydroxyl radicals in the glycol might reduce some Bi3+to form metallic Bi,and the plasma interaction of Bi could yield a large fraction of electrons.Photogenerated electrons on α-Bi2O3could be accreted at the Fermi energy level of Bi rather than complexed with holes (Fig.21(g)).

Fig.22. (a) Synthesis diagram of Bi5O7I-100 and Bi5O7I-001.Reproduced from Ref.[169] with permission of American Chemical Society,copyright 2016.(b) Photocatalytic nitrogen fixation efficiency of Fe-BMO with different iron content.(c) Diagram of the photocatalytic process envisaged for 0.5% Fe-BMO.Reproduced from Ref.[170] with permission of Elsevier,copyright 2019.(d)Synthetic pathway of Bi@BOB-BMO ternary heterostructures.(e)Mechanism of nitrogen fixation by Bi@BOB-BMO-2.Reproduced from Ref.[171]with permission of Elsevier,copyright 2019.(f)TEM image of BOC/OV3 and potential mechanisms for the N2 fixation process.Reproduced from Ref.[172]with permission of Elsevier,copyright 2021.
Bismuth oxygen halides (BiOX,X=Cl,Br and I) as a family of layered ternary oxides are prone to generate vacancy by being separated by two halogen atomic plates interacting through intermolecular forces to form a layered structure comprising [Bi2O2]2+plates.It also has an indirect band gap which suppresses the combination of optically generated electrons and holes [198].Bismuth halide with an open layered crystal structure possesses an outstanding photocatalytic performance,and has better photovoltaic ammonia ability than titanium dioxide under UV light irradiation.Quasi 2-D materials are featured by a high ratio surface area and a large quantity of uncoordinated surface atoms exposure and can briefly evade from the lattice so as to form vacant space [81,198].Miet al.[198]acquired bidimensional square BiOI nanosheets possessing high-reactive(0 0 1)facets by the hydrothermal process.In comparison with microporous plates,2D similarly square BiOI nanosheets exposed about 93% of the high reactive (0 0 1) facets,whereas BiOI microporous plates exposed a lower percentage of about 85%.The BET showed that 2D square BiOI nanosheets had triple exposed (0 0 1) facets compared to BiOI microporous plates in the same volume space.The 2D square BiOI nanosheets were featured by relatively small band clearance which exactly equivalent to the visible light range,so as to produce more electronhole pairs.Simultaneously BiOI nanoflakes possessing a more exposed (0 0 1) facets could generate more internal electrostatic fields,which were induced to enhance the splitting performance of photo-induced hole-electron pairs.Bi@BiOBr microspheres self-assembled by countless two-dimensional (2D) interspersed BiOBr nanosheets form blossom-like morphologies with oxygen vacancies,exhibiting remarkably enhanced photocatalytic performance for solar nitrogen fixation [168].The photocatalytic N2fixation experiment was all undertaken at ambient temperature with a 300 W point light source and it was found that Bi@BiOBr,which was processed in hydrogen at the 200°C,had the maximum ammonia production efficiency (181.2 μmol∙g-1∙h-1) compared to the material at other temperatures.This enhanced photocatalytic capability was attributed to that the material possessed a suitable bismuth metal content,the abundance of oxygen vacancies and a better BET specific area,which enhanced visible light absorption and photo-induced charge separation.pH is functioned as a criterion approach to control the Bi based photocatalyst surface.However,there are some BixOyXzthat are inherently produced by pH adjustment so that they cannot be adapted to the exposed planar surface by changing pH.Bi5O7I was less efficient at separating photoinduced carriers.Bi5O7I-100 was obtained by dispersing few precursor in deionized water,followed by a simple hydrolysis process after heat treatment [169].Bi5O7I-001 complex was obtained by direct calcination of the precursor (0.5 g) at 400°C for a long time(Fig.22(a)).20% methanol was used as sacrificial reagent for N2immobilization on Bi5O7I.Bi5O7I-001 (111.5 μmol∙L–1∙h-1) had a slightly higher photocatalytic ammonia generation rate than Bi5O7-I-100 (47.6 μmol∙L–1∙h-1),with less reduction in activity after cycling a few times.DRS,MS and VB-XPS showed a negative CB position for Bi5O7I-001,which implied that the photocatalytic property was dependent on the exposure of the 001-cut surface.
Other than that,Bi2MoO6and Bi2O2CO3have also been investigated[13].A Fe-doped Bi2MoO6for effective photocatalytic N2fixation was prepared [170].The rate of the optimum photocatalytic capability could attain 106.5 μmol∙g-1∙h-1,which was 3.7 time more than that of pure Bi2MoO6(Fig.22(b)).Fe doping increased the surface area of the catalyst as well as enhanced the absorbance of the catalyst.The photogenerated holes moved to the surface to react straight away with the hydroxide adsorbed on the surface to produce O2and water,while a fraction of the electrons would react directly with N2to produce ammonia (Fig.22(c)).Bi@BiOBr-Bi2MoO6,a new ternary heterostructure photocatalyst which has a homogeneous round-shapes structure with diameter of about 200 nm,was obtained from a solvent thermal synthesis by Lanet al.(Fig.22(d)) [171].The prepared hetero-junctions catalyst exhibited excellent photocatalytic activity,showing that the Bi@BOB-BMO-2 sample had outstanding NH4+production velocity of 167.2 mmol∙g-1∙h-1,above 2 times than that of both pure Bi2-MoO6and Bi@BiOBr.Bi@BOB were stacked microspheres with wrinkled surfaces,and the balls were assigned by a handful of thin nanosheets that inserted various holes between them.Regular comparatively smaller BMO nanosheets with higher crystallinity were sitting on the surface of Bi@BOB which still maintained the overall spherical structure but with smaller sphere size and enhanced dispersion.Bi—O bonds in the distinctive lamellar of BiOBr interacted with reduced glycerol during synthesis to produce oxygen vacancies,which enhanced the absorption in the visible region.On visible light illumination,the photo-excited electrons would travel from the CB of Bi@BOB to BMO.At the simultaneous time,BMO would also undergo light activation irradiation and the photo-generated electrons would travel to CB,afterwards to link with the waiting N2to get NH3(Fig.22(e)).Fenget al.[172]demonstrated that the OV embellished nano-flake material Bi2O2CO3i.e.BOC/OV displayed an extraordinary reaction property under visible beam(Fig.22(f)).The superficial oxygen vacancies offered enriched active adsorption points for start-up of molecular N2,and the highestyield of 1178 μmol∙L–1∙g-1∙h-1was obtained for BOC/OV3 among all the prepared samples.The subsurface OVs could function as an E-trap to conjugate abundant localized electrons.Furthermore,the Fermi energy level was also projected to be displaced along with the increase of donor density and enhanced separation of photogenerated charges,providing more active sites,and thus improving the photocatalytic nitrogen fixation capacity of the BOC/OV samples.The NH+4yield firstly increased and then decreased with the increase of the OVs concentration.This was attributed to the fact that OVs with too high concentration could also serve as a complex center of charge and inhibit the migration of free charges.At present,the photocatalytic activity of bismuthbased catalysts as before needs to be improved.
5.3.Other emerging catalysts
Intermediate metal sulphones have garnered widespread attention as a photocatalyst with a narrow-forbidden band.In 1980,Miyamaet al.[199] successively developed CdS/Pt duplex silica flake composite material for the inhomogeneous photocatalytic ammonia production,and the obtained NH3yields were better than those of pristine CdS,and however the photocatalytic activity was still very low.Afterwards,the photocatalytic synthesis of ammonia was realized under illumination by employing the CdS/Pt/ RuO2complexes which can drive the preliminary reaction of N2,while the process of electron capture in the CB of the semiconductor CdS was carried out with the participation of RuO2.Cd0.5-Zn0.5S was adopted for the photochemical NH3generation,and the ammonia generation rate of Cd0.5Zn0.5S with loaded Ni2P was 101.5 μmol∙g-1∙h-1in deionized water without scavengers(Fig.23(a),(b)) [173].The results indicated that Ni2P as a supplementary catalyst dramatically facilitated the photogenerated electron and hole transfer.Motivated by oxide photocatalysts,metal sulfide photocatalysts with enriched S Vacant space(Vs)have also been applied to light-derived ammonia preparation.A family of ternary metal sulfides possessing Vs,e.g.,Zn0.1Sn0.1Cd0.8S and Mo0.1Ni0.1Cd0.8S,were obtained.The concentration of N2was reduced in visible light,while the concentration of sulfur vacancy which can capture electrons was linearly related to the ammonia yield (Fig.23(c)) [186,200,204].The efficiency of ammonia synthesis of ultrathin molybdenum disulfide was as high as 325 μmol∙g-1∙h-1,whereas bulk molybdenum disulfide had no capability to reduce N2under the same conditions,which was attributed to the fact that ultrathin molybdenum disulfide nanosheets could yield light-induced deletions with high concentrations of localized electrons,facilitating the multi-electron process of N2photo immobilization [174].The BiVO4/Sv-Zn2S4(BVO/Sv-ZIS) containing a large number of sulfur vacancies was applied for photocatalytic ammonia synthesis [175].Among the catalysts,the two phases were in close contact and formed Z-scheme heterojunctions with BiVO4as the core and ZnIn2S4nanosheets as the core–shell layer,which facilitated the light-generated electron shift from BiVO to the conduction band of ZnIn and improved the enrichment of light-generated electrons on ZnIn surface (Fig.23(d)).In addition,the sulphur vacancy 2S4 nanosheets abounding on the Zn surface were very suitable for the adsorption of inert nitrogen and activation of two molecules.Also,they were used as the trap for photo-derived ammonia production to boost the activation efficiency.The complex of nitrogen fixate is made up of two proteins: ferritin,which is responsible for providing the electron,and Mo-Fe-protein,which utilizes the supplied electrons to reduce N2to ammonia,stimulating the researchers to study the catalytic behavior of the synthetic complexes of Fe,Mo and S and to engineer organ sulfide catalysts to enhance the N2fixation activity [145,146].
Nitrilase MoFe-based proteins were separated from nitrogenfixing bacterium and subjected cadmium sulfide (CdS) nanocrystals to photo-sensitization of the nitrilase molybdenum-iron(MoFe)protein in this process by Brownet al.[201],in which light capture substituted for ATP hydrolysis to push cracking of N2to ammonia(Fig.23(e)),and under light exposure at 405 nm(comparable to CdS nanorods’ bandwidth),the biocomponent lightcatalyst units exhibited impressive ammonia synthesis photocatalytic activity,with an ammonia formation speed of approximate 315 nmol∙mg-1∙min-1,TOF of 75 min-1and 63% of the ATP coupling reaction velocity of the enzyme complex.The authors concluded that it was the possible key that the continuous fast transmission of electrons resulted from the intensive photo absorption of CdS nanorods,allowing the realization of FeMo-co state with four-electron reduction.The MoFe based proteins were replaced by ‘‘Mo2Fe6S8-Sn2S′′6copper gels composed of Mo2Fe6S8(SPh)3and Sn2S6ligands (Fig.23(f)),where the Sn2S6copper gel served as a light collector[203].Copper gels demonstrated promising photocatalytic stability over the 72-h test period.These mimetic sulfur gels possessed enhanced light absorption and high-level surface area,as well as favorable water stability,allowing ambient photo driven nitrogen fixation.Similarly,biomimetic chalcogen enzymes with bicube [Mo2Fe6S8(SPh)3]and monocube(Fe4S4) were explored for their unique structure (Fig.23(g))[204].In the study,Mo-free sulfur gels only containing[Fe4S4]clusters were found to be more efficient in predriving ammonia synthesis than sulfur gels including [Mo2Fe6S8(SPh)3],which showed that Mo was non-imperative for the conversion of N2to ammonia.Therefore,the authors concluded that the reactive site in mimetic sulfur gels was distinct from the active site in nitrogen fixation enzymes.
Graphite carbon nitride(g-C3N4),a one-class non-metallic polymeric semi-conductor,have tremendous photocatalytic NRR activity with various advantages such as inexpensive,high abundance,brilliant absorption of visible light,excellent stoichiometric and photochemical resistance [13].The primitive g-C3N4normally has a relatively low specific surface area,unfavorable for N2shift and the complex annihilation rate of light-generated electrons and vacancies,leading to inefficient photocatalysis.g-C3N4and Fe(NO3)3∙9H2O dispersed in ethanol and the iron trioxide packed with perforated graphite-carbon-nitride was prepared [176].When Fe content of the sample was less than 1% (mass),hybrid catalyst reduced nitrogen to ammonia with an ammonia yield of 47.9 mg∙L–1∙h-1,which was 6 time of bare g-C3N4.The iron trisulfide particles were arranged as an aggregation on g-C3N4sheet surface and the aperture was not uniform.The oxidation and etching of the g-C3N4surface by iron trioxide resulted in an increment of its superficial area and porosity bulk with a decrease of about 3%in nitrogen content.Fe2O3/g-C3N4hierarchy for photo-oxidation of ethanol and reduction of N2was probably based on a z-type mechanism (Fig.23(h)): The VB of g-C3N4was proximate to the CB of iron trioxide,and the photogenerated electrons in the CB of iron trioxide were comparable to those in the VB of C3N4,which rapidly reorganized and enhanced the separation of photoelectrons and photo holes.This z-solution system was capable of maintaining a strong redox capacity,which strengthened the photocatalytic property.On the other hand,this photo-catalytic system had a large exposed surface area and extensive mesoporous structure,which promoted the adsorption of N2and facilitated its further conversion to ammonia.

Fig.23. (a)The reaction process of ammonia production by Cd0.5Zn0.5S.(b)Ammonia rates of Cd0.5Zn0.5S and Ni2P/Cd0.5Zn0.5S under visible light irradiation.Reproduced from Ref.[173] with permission of Elsevier,copyright 2017.(c) Variation of nitrogen fixation performance of prepared catalysts with sulfur vacancy concentration.Reproduced from Ref.[200]with permission of Royal Society of Chemistry,copyright 2016.(d)Schematic diagram of the formulation of the Z-type electron transition mechanism of ZIS and BVO.Reproduced from Ref.[175]with permission of Elsevier,copyright 2022.(e)Reaction program for the reduction of N2 to ammonia by MoFe protein biohybrids and the reaction process of substitution with cadmium sulfide.Reproduced from Ref.[201] with permission of Science,copyright 2016.(f) Diagram of Mo2Fe6S8-Sn2S6 bionic sulfur gel synthesis.Reproduced from Ref.[202] with permission of American Chemical Society,copyright 2015.(g) Reaction roadmap for the synthesis of different biomimetic sulfur gums.Reproduced from Ref.[203] with permission of PNAS,copyright 2016.(h) Schematic diagram of photocatalytic N2 reduction with iron trioxide loading.Reproduced from Ref.[176] with permission of Elsevier,copyright 2019.
The exposed active N atoms were available to hydrogenate and promote the synthesis of ammonia in the process,those naked N atoms might be securely anchored by forming B≡N≡C ligands in the nanosheets(BCN) [177].Additionally,the B≡N≡C coordination formed in g-C3N4not only validly enhanced seeable light trapping as well as suppressed the reconstitution of photosynthetic operators in g-C3N4,but also served as a catalytically active site for the whole conversion process.The obtained BCN manifested an ammonia output of 313.9 μmol∙g-1∙h-1,which was virtually ten folds than that of the pristine g-C3N4(Fig.24(a)).Converted 2D g-C3N4laminate with modified ultra-thin flakes of MnO2–xwas used as a nitride-fixing photocatalyst [178].Aiding in the performance of MnO2–x,the ammonia production was achieved at a rate of 225 mmol∙g-1∙h-1,which exceeded more than twice that of the primordial 2D1-C3N4(107 mmol∙g-1∙h-1).The ultra-thin sheets of MnO2–xnarrowed the clearance between the vehicles and the photocatalyst surface,increasing the amount of electron transfer.SEM image (Fig.24(b)) clearly revealed the graphite-like ultrathin structure of MnO2–x/g-C3N4with convoluted surface.XPS showed the emergence of C-O species on the surface at 286.4 eV,predicting that the ultrathin flake MnO2–xwas also chemically bonded to 2D g-C3N4.A redox ring was formed between MnO2–xand 2D g-C3N4,which consumed electrons in the former and hollows in the latter,thus facilitating the detachment of the optical carriers of 2D g-C3N4.A volcano plot (Fig.24(c)) demonstrated that the 1% MnO2–x/2D g-C3N4sample possessed best NRR property.Too much MnO2–xsurface coating,which impeded the optical absorption ability of 2D g-C3N4,caused such a situation that as the cycle number increased,some MnO2–xcould not form a chemical bond with 2D g-C3N4during the hydrothermal reaction and fell off,leading to a persistent activity decrease.The high emission intensity of 2D g-C3N4was found by PL spectroscopy,illustrating the high complexation rate of optical electron-hole pairs,which suggested that the introduction of ultra-thin sheets of MnO2–xeffectively suppressed the complexation.The vacancies on g-C3N4oxidized Mn3+to produce NH3,while the photogenerated electrons on MnO2–xcontributed to the generation of Mn3+(Fig.24(d)).In addition to co-catalyst amendment,xenobiotic junction structures have also emerged in the exploration of sophisticated photocatalysts for ammonia production owing to their exceptional properties.From synthesis method by Fig.24(e),TiO2@C/g-C3N4heterojunctions sourced from MXenes also received attention[179].The structure showed enriched surface defects,relatively large electron supply capacity,desirable light harvesting ability,superior charging ability and enhanced nitrogen activation.The ammonia production rate was as high as 250.6 μmol∙g-1∙h-1under visibility lighting due to the unique structure.It was detected that the photocatalytic capability of g-C3N4and TiO2@C heterojunctions was superior to that of TiO@C+g-C3N4samples prepared by mechanical mixing (132.3 μmol∙L–1∙g-1∙h-1).For example,in the mixed g-C3N4/ZnMoCdS system(Fig.24(f))[206,207],the interaction of the intense electronic pair between the two components promoted the stripping of the stimulated electrons and holes through the interfacial charge.Under visible light excitation,a higher rate of ammonia production was produced than either component alone.

Fig.24. (a) The comparative photoproduction yields of ammonia from CN and BCN photocatalysts.Reproduced from Ref.[177] with permission of Wiley Online Library,copyright 2020.(b)SEM image of 1%MnO2–x/2D g-C3N4.(c)Comparison of ammonia synthesis for different loading samples.(d)Mechanism of NRR process envisioned for g-C3N4.(e) Schematic illustration of the composition process of TiO2@C/g-C3N4.Reproduced from Ref.[178] with permission of Elsevier,copyright 2020.(f) Diagram of the energy band structure and electron-hole separation of TiO2@C/g-C3N4.Reproduced from Ref.[179] with permission of Royal Society of Chemistry,copyright 2018.(g)Schematic diagram of a polyhedron with an ultrathin LDH structure of molybdenum hexahedra.(h)Schematic diagram of photocatalytic N2 fixation process.(i)Comparison with the yield of each control group of the experiment.Reproduced from Ref.[205]with permission of Wiley Online Library,copyright 2017.(j)Ammonia yields of different ZnCr-LDH samples irradiated with UV–Vis light (engraftment with irradiation time).Reproduced from Ref.[180] with permission of Wiley Online Library,copyright 2020.
LDHs trespass into the field of photocatalytic ammonia production since their metal cation composition and easily tunable thickness,allowing doping defects and changing band gap size,LDHs are made of MO6octahedra with shared edges (Fig.24 (g),(h)) [205].By forming LDH nanosheets with few thicknesses,OV can be produced the on the surface or edges of the nanosheets.As indicated in Fig.24(i),it was found that extra-thin nanoflakes of CuCr-LDH demonstrated excellent photo-catalytic ammonia yield without any sacrificial reagent under visible light in water.This outcome was attributed to the twisted configuration of the super-thin LDH nanolaminates and the compression strain generated by the large amount of oxygen flaws,which substantially promoted the chemical transformation of N2.Zhaoet al.[180]reported the generation of LDH-NSs,ZnAl-LDH and NiAl-LDH with a large quantity of O vacancies,cationic vacancies as well as unsaturated metal sites by an alkali etching process.All the alkali-etched photocatalyst manifested superb photocatalytic ammonia production activity compared to the unetched LDHNSs.Among them,the photocatalytic ammonia production activity of etched ZnCr-LDH was 33.19 mmol∙g-1∙h-1with an AQE of 0.11%at 550 nm,1000%larger than that of the unetched ZnCr-LDH (Fig.24(j)).
A facile hydrothermal approach was used to build a family of novel NiFe-LDH-derived sulfide ([NiFe]S) micelles (Fig.25(a))[181].Strikingly,the ammonia production rate of[NiFe]S catalysts reached 111.83 μmol∙L–1∙h-1under visible lighting in water,which was around ten folds better than that of NiFe-LDH microspheres(Fig.25(b)).The explanation for this was the high opticalinduced isolation and delivery rates of [NiFe]S microspheres.The diameters of NiFe-LDH microspheres were found to be in the range of 8–10 μm by SEM.But after sulfidation,the microspheres were clumped up slightly and the microsphere surface was cleaved.The resulting interfacial Ni—S bond and Fe—S bond were identified by XPS spectra,which were conducive to the effective transmission of photogenerated electrons.The pure NiFe-LDH microspheres were detected in the UV–Vis DRS diffuse reflectance spectra and only absorbed a large amount of UV light.After being etched with thioacetamide,the resulting [NiFe]S-2 microspheres were redshifted at the absorption edge,indicating that the prepared[NiFe]S-2 microspheres exhibited improved visible light responsiveness and thus enhanced photocatalytic N2immobilization activity.The remarkable decrease in charge resistance derived from electrochemical impedance spectroscopy (EIS spectra) was a proof that the quick isolation rate of light-generating carriers and effective interfacial charge shift were obtained in the [NiFe]S-2 microspheres.Besides,the photocatalytic ammonia generation rates of [NiFe]S-2 microspheres at different temperatures coincided well with the Arrhenius equation(Fig.25(c)).After five cycles of experiments,the photocatalytic ammonia generation rate decreased from 111.83 to 96.75 μmol∙L–1∙h-1,with only about 13.48% reduction.Its catalytic process could be summarized as(Eqs.(5)–(10)):
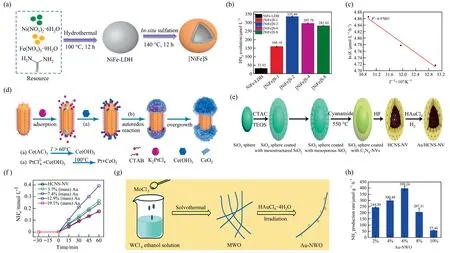
Fig.25. (a) Preparation step diagram of NiS microspheres.(b) Ammonia production activity of [NiFe]S microspheres prepared from NiFe-LDH and different content of TAA feedstock.(c) Curves of ammonia production rates at different temperature regimes.Reproduced from Ref.[181] with permission of Springer,copyright 2021.(d) The synthesis process of an Au/endCeO2 nanostructure.Reproduced from Ref.[182]with permission of American Chemical Society,copyright 2019.(e)Schematic representation of the synthesis process of Au/HCNS-NV nanostructures.(f)Effect of the Au loading amount on the photocatalytic ammonia production rate.Reproduced from Ref.[183]with permission of Royal Society of Chemistry,copyright 2020.(g) Preparation scheme of Au-MWO.(h) Rates of photocatalytic ammonia generation from Au-MWO doped with different Au doping concentrations.Reproduced from Ref.[184] with permission of Royal Society of Chemistry,copyright 2021.
Localized surface plasmon resonance denotes the strong interplay of a plasmonic nanostructure with incident appropriate radioactivity,leading to the resilience of surface-conducting electrons to excite positive nucleon masse.Extensively noted for the creation of thermo-electronics(i.e.,high-energy electrons),plasma nanostructures produce hot electrons and holes upon light absorption,which can be transferred to the reacting moleculeviaa variety of pathways.Stimulated thermo-electrons enable excitation to drive reduction reactions and possibly reinforce the heterogeneity of surface charges,thereby boosting reaction rates [208].In addition to the Au-doped class of photocatalysts with oxygen vacancy-driven N2adsorption and activation of TiO2groups mentioned in the previous sections,there still exist a number of CeO2-based materials with similar catalytic ability to TiO2,which presents excellent catalytic ability because of the widespread availability of OVs at their surface.In the existence of a small amount of K2PtCl4,cerium oxide was selectively evolved at the end of gold nanorods.As shown in Fig.25(d),the deposited CeO2was crystalline and consisted of a great density of OVs and K2PtCl4was used to activate the autocorrelation reaction with the CeO2precursor to selectively grow the semiconductors at the terminal end of gold nanorods by preferential adsorption[182].The ammonia yield of Au/end-CeO2was 114.3 μmol∙g-1∙h-1,which was 6.2 times higher than the Au-CeO2core–shell nanostructure.Guoet al.[183] prepared hollow mesoporous carbon nitride spheres(HCNS)embedded with gold nanoparticles of different thicknesses,using the process shown in Fig.25(e).Under visible light illumination,the NRR activity reached a maximum of 783.4 μmol∙g-1∙h-1at a gold loading of 7.4% (mass) (Fig.25(f)).The NVs on the chemosphere surface of HCNS-NV adsorbed unfettered N2,which were also the target of the photogenerated electrons of HCNS-NV itself and the trapping sites of the gold nanoparticle plasma excitation.The electrons on the adjacent carbon atoms of the NV sites were transferred to the trapped N2molecules,causing the engagement of the N≡N bond,which was diminished by the trapped photogenerated electrons.The gold nanoparticles markedly improved the carrier separation efficiency and promoted the concomitant happening of redox reactions.The authors therefore concluded that NVs in HCNS served as activation locus for N2,catching Lightexcited electrons of HCNS and resonating with protons of embedded gold nanoparticles to reduce nitrogen to ammonia.Mo-doped W18O49nanowires (Au-MWO) moored by plasmonic gold nanocrystals were synthesized by the method shown in Fig.25(g) [184].Mo5+preferred to replace W5+species at the surface defect sites instead of creating new flaws.Mo doping failed to change the crystalline phase of WO.Au nanoparticles were homogeneously distributed and there existed electron shift between MWO and Au nanocrystals.In accession to enhance nitrogen adsorption and activation,the moo-doped Au nanocrystals not only protracted the light absorption to generate high-energy electrons,but also lowered the desorption energy of NH,which was beneficial to the release of active sites and further enhanced the catalytic reaction of nitrogen fixation.Among them,N2-saturated water and methanol were used as the precursor and sacrificial agent,respectively.WO progressively enhanced the rate with the introduction of molybdenum,and MWO-2 showed the maximum level of N-fixation productivity up to 174.97 μmol∙g-1∙h-1.Fig.25(h) demonstrated the effect of Au filling to the properties of Au-MWO photocatalytic nitrogen fixation and exhibited an outstanding ammonia generation speed of 399.24 μmol∙g-1∙h-1as a result of the synergistic effect of molybdenum diversity and gold nano-crystal packing.The calculations expressed that the last step of ammonia desorption was regarded as the rate-limiting step of the overall nitrogen consolidation process.
6.Other Ammonia Synthesis Approaches
6.1.Chemical looping ammonia synthesis
The chemical looping process refers to splitting the target reaction into two or more step-by-step reactions,all of which can be carried out in different spaces,times or under diverse reaction conditions,and the step-by-step reactions can be optimized one by one to achieve the overall target reaction.In the chemical looping process,a certain reaction carrier,undergoing consumption and regeneration throughout the chemical cycle,can be used for the transfer of oxygen,nitrogen,carbon or hydrogen species depending on the target reaction[15].Chemical looping processes,involved in gas–solid or liquid–solid interface chemical reactions in nature,are closely related to heterogeneous catalytic processes so that some heterogeneous catalysts can also be utilized as reaction carriers in chemical looping processes.There are many differences,in that chemical looping takes a step reaction means with various feed times and distinct reaction temperature and pressure independent of conditions for N2activation and ammonia generation,thereby bypassing the H chemo-sorption of reactive substances present on the catalyst surface [2].As illustrated in Fig.26,according to the different forms of ammonia release by nitrogen carrier,the chemical looping synthesis process of ammonia can be divided into chemical looping with water as hydrogen source (expressed as H2O-CL) [210],chemical looping process with hydrogen as hydrogen source(expressed as H2-CL)[211]and chemical ring mediated using the alkali/alkaline earth metal hydride and imide(expressed as AH-CL) [209].The relatively significant properties of chemical looping ammonia synthesis methods are concluded in Table 8.
The most characteristic H2O-CL is the AlN-Al2O3chemical looping process.Steinfeldet al.[210,217]designed a two-step chemical looping synthesis of ammonia powered by solar thermal collectors(Fig.27(a)).In the nitrogen fixation process,Al2O3undergoed a reaction with a reducing feedstock such as methane or carbon in a nitrogen atmosphere to form AlN and CO;AlN was hydrolyzed with water at high temperature to produce ammonia and AlN back to Al2O3.In terms of the thermodynamic perspective,the first step of the reaction is a strong heat uptake process,which requires high temperature for the reaction to occur and the second step is an exothermic reaction,which is reflected in the following Eqs.((11)-(13)).
Ammonia production: 2AlN+3H2O →2Al2O3+2NH3

Fig.26. Simple diagram of the three Chemical looping cycles H2O-CL and H2-CL and as AH-CL.(M=Al,Cr,Mn etc.) Reproduced from Ref.[209] with permission of Nature Publishing Group,copyright 2018.
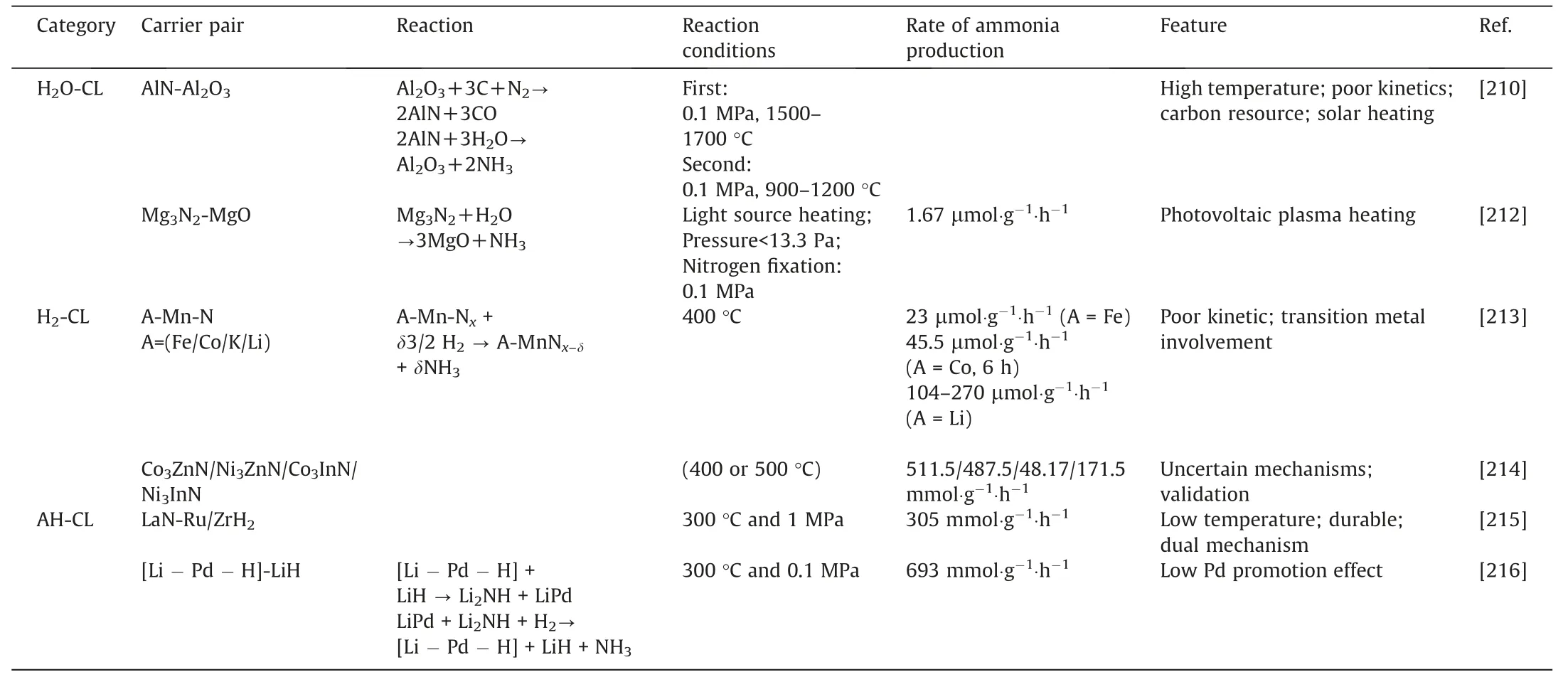
Table8 Summary of typical and new chemical looping ammonia synthesis performance analysis
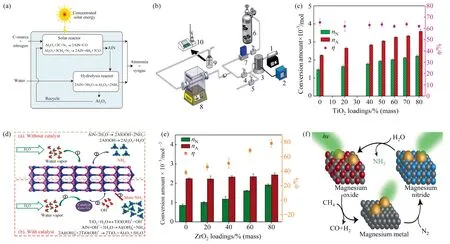
Fig.27. (a) A two-step process scheme for the production of ammonia in a solar thermochemical cycle.The first heat absorption step is to produce AlN.the second exothermic step is the steam hydrolysis of AlN to produce ammonia and reformed alumina.Reproduced from Ref.[209] with permission of Nature Publishing Group,copyright 2018.(b) Fixed bed reactor structure.(c) Ammonia yield (nN),conversion AlN(nA) and corresponding ammonia production efficiency (η) for different titanium dioxide loaded catalysts.(d) Potential routes for N2 detoxification reactions and ammonia production.Reproduced from Ref.[218] with permission of Elsevier,copyright 2018.(e) Ammonia yield (nN),conversion AlN(nA) and corresponding ammonia production efficiency (η) for different zirconium dioxide loaded catalysts.Reproduced from Ref.[219]with permission of Elsevier,copyright 2020.(f)A schematic illustration of the chemical cycle of ammonia synthesis of Mg metal.Reproduced from Ref.[212]with permission of American Chemical Society,copyright 2019.
This AlN-Al2O3process is characterized by the inexpensive raw material Al2O3,ambient pressure operation,solar thermal collector energy supply,no CO2emission,and co-production,etc.The disadvantages are that the high temperature conditions are demanded for both steps,1500–1700 °C for the first step and 900–1200 °C for the second step,resulting in high energy consumption and relatively high requirements for the reactor equipment.In addition,the decomposition of ammonia into nitrogen and hydrogen is thermodynamically spontaneous reaction at high temperatures,which leads to a lower yield of ammonia [15,210,217].In order to optimize the reaction kinetics,researchers have tried multiple additives or catalysts.For the purpose of nitrogen fixation reaction,it was found that some Ca-containing compounds such as CaF2,CaCO3,Ca(OH)2and CaC2formed aluminates with Al2O3at 1350 °C,and the aluminates could be dissociated by gasification at high temperature to produce aluminum vapor for rapid nitration[218].TiO2was also explored in classical fixed bed reactor(Fig.27(b)),and water had excellent dissociative adsorption properties on the surface of titanium dioxide,whereas,the liberated hydroxyl group (OH–) had a vital function in the N2desorption.The aluminum nitride transformation and ammonia output were found to increase as titanium dioxide was loaded in the reactants and the authors gave potential routes for N2detoxification reactions and ammonia production (Fig.27(c),(d)).From Fig.27(e),it is concluded that zirconium dioxide likewise enhances the formation of ammonia because molecular adsorption of ammonia occurs on the zirconium dioxide surface,thus preventing the decomposition of ammonia [219].Sweareret al.[212] introduced a three-step ammonia synthesis process utilizing the photothermal conversion of TiN which was a plasmonic material (Fig.27(f)): TiN nanoparticles were exposed to light irradiation to generate heat to drive the reduction of MgO to metallic Mg by CH4and to produce CO and H2in a 1:2 ratio,which could be used as a substrate for Fischer-Tropsch synthesis.The second step was the nitration of magnesium metal in a nitrogen atmosphere to produce Mg3N2.Finally,Mg3N2was hydrolyzed to produce ammonia,which regenerated magnesium oxide and finalized the cycle.Since only a restricted surface of the material would be subjected to light,the rate of ammonia production was low,about 1.67 mmol∙g-1∙h-1.In considering to increase the rate of ammonia production from the photothermal coupled chemical looping,it was recommended to enlarge its photothermal conversion efficiency or try other isoexcited materials [15].
The lattice nitrogen may discharge during the reaction and recharge during the 2nd stage at various reactive environments,thus mitigating the thermodynamic restrictions.A-Mn-N (A may be Fe,Co,K,Li)substances were acquired by a regular precipitation calcination method with amido sodium as raw material.The presence of Mn3N2restricted the reactivity of the released lattice nitrogen,only 3.1% MnN2reacted with hydrogen to get ammonia,and the majority of the crystalline lattice nitrogen was removed in the form of N2.Nevertheless,the existence of co-metals probably contributed to the nitrogen delivery performance of manganese nitride.Application of H2direct reduction to produce ammonia was identified (A-Mn-Nx+δ3/2H2→A-MnNx–δ+δNH3) and the usefulness of the doping metal was evaluated: Fe hastened the consumption of lattice nitrogen in the form of N2,rather than the reaction with H2[213].
The reaction of H2-CL mainly exists on nitrogenous compounds.Mn has been the bursting point of research for ammonia production by H2-CL on account of its large content and affordability.Solid manganese grains are modified into Mn nitride by exposure to N2gas at atmospheric pressure and high temperature,followed by the activation of hydrogen on the solid surface and release of N atoms from the solid to produce ammonia[220].However,the modification of Mn nitride has become the follow-up research direction because of the inferior kinetic properties of Mn nitride [15,220].Doping of Ni in the superficial surface layer improves the reactivity by virtue of its adsorption capacity of H [221].The oxidation of superficial Mn is achieved by adding NaOH,along with a quantum leap in the yield and chemical cyclization kinetics of Mn [222].It was reported that the existence of Fe destroyed the Mn-N bond in Mn6N2.58and Mn4N,thus augmenting disintegration and leading to a lower ammonia yield in the reaction with H2.The results gained by the addition of cobalt were comparable to those of the Fe-containing system.The synthesis of ammonia was intensely suppressed by the addition of potassium to Mn3N2,oppositely,potassium acted as effective promoter of iron and rhenium-based catalyst.In comparison,a dramatic improvement in the migration property of nitrogen was achieved by lithium which was utilized as a co-metal.Some authors[213]proposed‘‘a dual active center catalyst‘‘in which the lithium was used to deplete Nadon the surface as well as initiate the hydrogenation process,whereas the Mn(N)platform reactivated N2in a consecutive way.Li-Mn-N (Mn/Li about 1:10) was intended to function as a nitrogen shift agent rather than a catalyst.
The AH-CL process involves the interconversion of alkali(earth)metal hydrides and iminomers for nitrogen fixation and ammonia production,as shown in Eqs.((14),(15)).In the process of nitrogen fixation,the alkali (earth) metal hydride interacts with N2to produce the corresponding amino compound,which then acts with H2to release ammonia and regenerates the hydride [2,211].
The nitrogen fixation process of hydrides is completed by the redox reaction of hydrides with N2,in which N2is reduced and the negative hydrogen species (H-) in hydrides lose electrons to form protons and H2.The nitrogen fixation and ammonia production temperatures of the hydrides are substantially lower than those of AlN and transition metal nitrides.It has been studied that nitrogen fixation of LiH,CaH2,MgH2,BaH2and other alkali (earth)metal hydride to produce the corresponding amino compounds or nitrides are all thermodynamically favorable reactions(Fig.28(a)).TMs with 3d orbital,such as iron,cobalt and nickel can be used as catalysts to significantly accelerate the rate of hydride nitrogen fixation and amino compound hydrogenation to produce ammonia in the order of Ni >Co >Fe.Ni has long been considered as a poorly active transition metal in the conventional synthetic ammonia catalytic process.The TM-AH composites outperform the Csfacilitated Ru/MgO 2–3 times and the optimized reaction conditions (1 MPa and 300 °C,even for Ni-Ba2H to produce ammonia at less than 100°C and atmospheric pressure)can be achieved substantially.In the AH-CL process,AH2has a decisive role in the activation of N2and in the conversion of the existence of the H valence form,while TM,as electron donors,not only acts as N2carriers but also catalyzes the formation of imides by the fixation of N2through hydrides[211].The augmentation of the catalytic activity of Mn4N by hydrides is BaH2>LiH>KH>CaH2>NaH[223].In the Ni/BaH2participation in ammonia production,BaH2was engaged in the conversion to BaNH in the N2reaction step.The H2phase was the opposite of the above,but might be accompanied by the formation of an intermediate phase (N-deficient barium imide),which was an incomplete hydrogenation step that implied that the latter is more dispersed than in the former [224].
The LaN-Ru/ZrH2catalyst had a very rapid rate of ammonia synthesis (up to 305 mmol∙g-1∙h-1) 350 °C at 1 MPa and was notably durable [215].LaN not only impacted the displacement of Ru species but also provokes the movement of electron density from La to Ru and finally to N2,thus contributing to the activation and hydrogenation of N2to *N2H2.The N in LaN reverted with the H in ZrH2to form ammonia,which could replenish N and H vacancies under atmosphere.Fenget al.[225] also found that the efficiency of the multifunctional composite carrier Mn2N-BaNH composed of manganese nitride and alkali or alkaline earth metal acylides was an order of magnitude greater than that of pure BaNH and Mn2N.
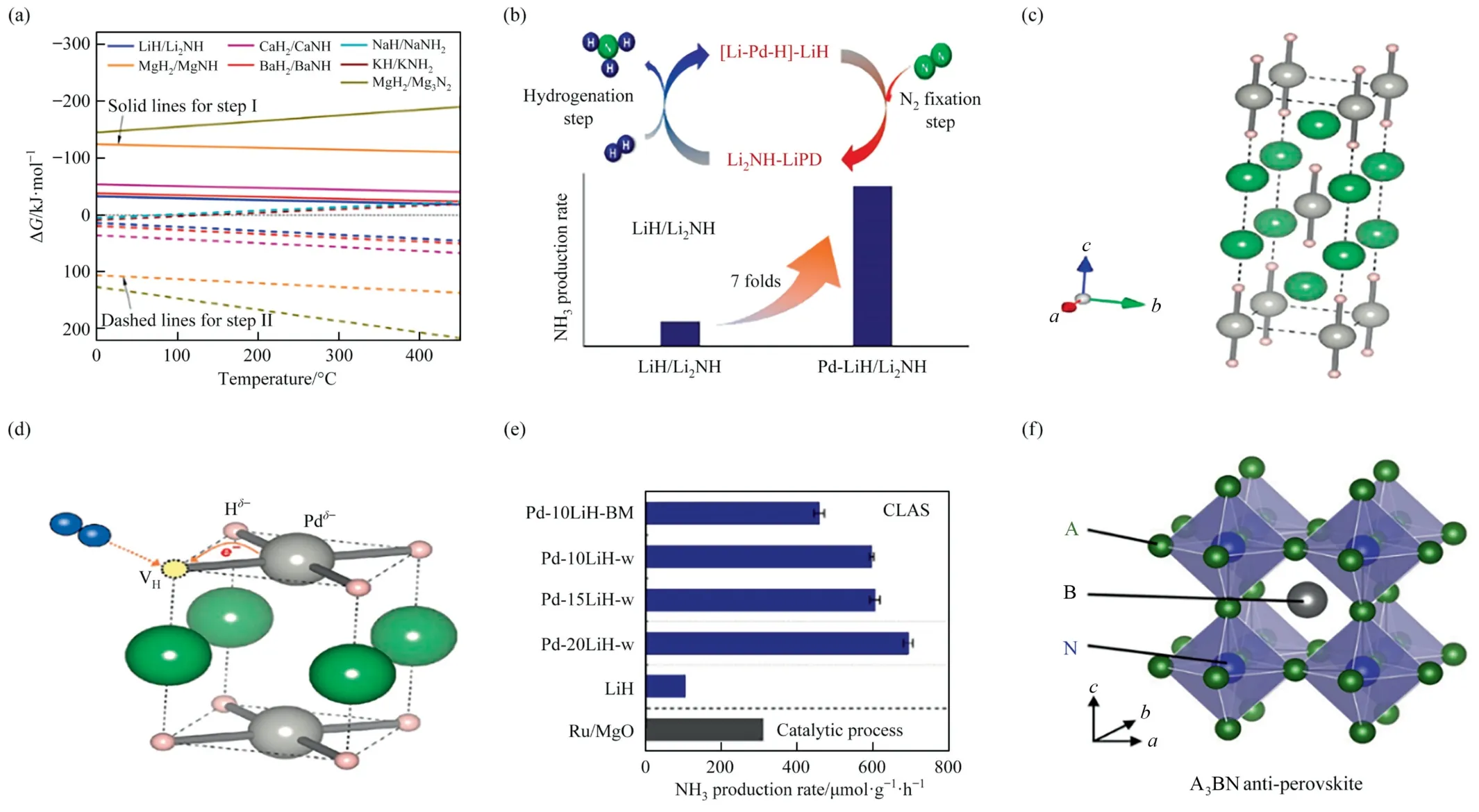
Fig.28. (a) Thermo-mechanical in AH-CL for steps I and II.The firm line is the temperature dependence of ΔG on hydride nitration.The dotted line is the temperature dependence of ΔG on the hydrogenation of the acyl imide.Reproduced from Ref.[209] with permission of Nature Publishing Group,copyright 2018.(b) Enhancement of chemical cyclic ammonia synthesis by palladium on lithium hydride and a sketch of the reaction.(c)Three-dimensional Li2PdH2 structure with green for Li,grey for Pd,pink for H and blue for N.(d)Three-dimensional structure of LiPdH0.7 and potential processes for the up-activation of N2 in this structure.Color meanings are the same as(c).(e)Comparative ammonia production rates of Pd-10LiH-BM,Pd-xLiH-w and lithium hydride via the chemical cycle pathway at 0.1 MPa pressure.Reproduced from Ref.[216]with permission of American Chemical Society,copyright 2021.(f) Crystal structure of A3BN anti-calcite with space group Pm-3 m.Reproduced from Ref.[214] with permission of Elsevier,copyright 2021.
In accession to the above metal elements,Yanet al.[216]wondered whether Pd accelerated the efficiency of ammonia production from lithium hydride.Pt was excluded for consideration,as it was found to have N2dissociation and robust RHE reaction ability in the previous study.Many characterization methods revealed that Pt provided a stepping stone for ammonia synthesis to form lithium palladium hydride (LiPdH0.7and Li2PdH2).The availability of LiPdH0.7favored the formation of lithium hydride which produced Li2NH and LiPd from nitrogen fixation,and the subsequent hydrogenation of Li2NH and LiPd resulted in the regeneration of lithium hydride and [Li-Pd-H] (Fig.28(b)).At 300 °C and 0.1 MPa,the ammonia yield was 693 mmol∙g-1∙h-1,and at a given temperature and 0.1 MPa pressure,N2(30 ml∙min-1)or H2(30 ml∙min-1)was alternately introduced to the Pd-LiH sample.The Pd promoted the immobilization of N2by lithium hydride to form Li2NH.Li2NH was used as a nitrogen vehicle and LiPdH0.7was as a catalyst.As depicted in Fig.28(c),Li2PdH2was observed in the existence of linear[PdH2]2-complexes(Pd valency was zero).Square planar[PdH4]complexes existed in LiPdH0.7,in which nearly 30% of the H-site unoccupied and Pd and nearby H–might transfer electrons to the antibonding orbital of the N2molecule to activate N2(Fig.28(d)).The activity was influenced by the preparation method,the molar ratio of Pd and lithium hydride (Fig.28(e)),and temperature.The intense affinity of H on the surface of lithium palladium hydride in thermal catalysis might hinder the activation of N2,making it much less efficient than the CL process.The promotion effect of Pd was still lower than that of the common Fe,Co,and Ni metals.The TM-free mediated barium hydride(BaH2)activated N2through the insertion of hydrogen vacancies,creating multiple ligandunsaturated Ba sites and enabling the adjacent lattice hydridic hydrogen to convert N2to NHx viaextinction and protonation[226].The resistance of ammonia production from the chemical rings of the chalcogenide nitrides Co3ZnN,Ni3ZnN,Co3InN and Ni3-InN has been investigated [214].Monophasic crystals possessing the desirable inverse chalcogenide structure were obtained by ammonolysis of the corresponding precursors (Fig.28(f)),the efficiency of Ni3ZnN and Co3ZnN were comparable and approximated to 3000 μmol∙g-1,the efficiency of Ni3ZnN was much superior than that of Co3InN(289(μmol∙g-1)with slightly above 1000 μmol∙g-1.It was demonstrated by X-ray diffraction that Co3Zn(In)N disintegrated along with the removal of lattice N,whereas Ni3Zn(In) N was transmuted to Ni3Zn(In) by the intermediate stages.The content of In/N in the synthesized Co3InN was higher than the theoretical value,because the melting temperature of In was much quencher than that of ammonification reaction,causing partial loss during the synthesis.The liberation of lattice N was topologically regulated.Ammonia production from nitrides under H2/Ar atmosphere occurred by a Mars-Van Kleven-like mechanism.Disparity in chemo affinity for Zn and In could lead to differences in the rate of ammonia production.Thermo-gravimetric analysis measurements showed that Ni3ZnN and Ni3InN were reproducible.
6.2.Plasma-assisted ammonia production
Plasma is regarded as the 4th state of substance unlike the solid–liquid-gas state,comprising vapor ions,non-binding electrons and un-ionized neutral particles.Plasma temperature is expressed as electron temperature and ion temperature.They are categorized as high-temperature plasma (same temperature),or non-thermal (low-temperature) plasma where the temperature of the electron is above than the matter making up the gas phase,depending on whether they have the same temperature [12].The Birkeland-Ede process,in which the reactants undergo ionization of gases in an air arc at 3000 °C and other processes to produce nitrogen oxides with productivity of 4%,was one of the first plasma-related nitrogen fixation processes implemented back in the early 1900s [16].There are many different atmospheric pressure plasma structures,e.g.,sliding arcs,plasma jets,and dielectric barrier discharges (DBDs).The plasma approachviaDBD method has represented the majority of investigations thanks to the accessibility of handling and doping of catalytic materials.In addition,DBDs are reactors in which an atmospheric pressure plasma is generated by applying a voltage between two electrodes separated by quartz or other ceramics (Fig.29(a)) [227].The plasma does not contribute to the reaction in the ammonia system alone.It is considered that there is some kind of interaction between the plasma material and the catalyst surface.Other than that,there is process in which an intermediate transformed by plasma effect firstly and subsequent ammonia production by other means.

Fig.29. (a)Schematic diagram of the electroreactor process with DBD.(b)Comparison of the analytical properties of Ni/SiO2-IWI with other controls.Reproduced from Ref.[227]with permission of Elsevier,copyright 2021.(c) Simple diagram of ammonia synthesis in a radio frequency plasma reactor.(d)Comparison of the ammonia synthesis rate performance of the two ZIF crystals studied with other materials.Reproduced from Ref.[228] with permission of American Chemical Society,copyright 2021.
The plasma-induced catalyst modification can be achieved by generating hot sites on the catalyst interface,causing an increment in surface mobility and sintering of catalyst particles or an increase in the local thermal ramp,thus influencing the reaction kinetics[12].Studies have also been done on Ni/SiO2,W,ZIF,Co,Si and other catalysts.The catalytic activation of nickel nanoparticles braced on silica was studied by placing them in a DBD apparatus,in which nickel wires were used as outer electrodes and tungsten as inner electrodes [227].Ni/SiO2-SiO2catalysts were prepared by primary wet impregnation method and surface organometallic grafting.The average particle size of the catalysts was significantly smaller as well as more homogeneous after the reaction.This status quo was explained by the slump of micro paths in the constructions of catalysts as a result of high-energy particle collisions.It was anticipated that the M—N bonding reinforced the synergistic effect of plasma and catalyst.And the comparison of alumina and silica as substrates for ammonia synthesis was done.Alumina has the higher dielectric constant than silica,which means that most alumina has weak and mid-acid sites,whereas the adsorption of ammonia at these sites is not intense.The salient points of silicon dioxide include:(1)its lower resistivity results in a more stable and uniform plasma discharge.(2) It can resist sintering and provides higher metal dispersion.(3)It also has the property of easily adsorbing hydrogen from the gas phase.Thus,higher synthesis rates of pure silica were obtained under hydrogen-rich conditions.Ni-containing particles were likewise beneficial for the hydrogen adsorption behavior.It was also verified that nickel is an optimal cocatalyst for plasma-catalyzed ammonia synthesis.(Fig.29(b)).Although Ni is an inactive metal,the presence of size effect makes it the best catalyst for plasma-catalyzed ammonia synthesis.The radio frequency (RF) plasma-assisted ammonia synthesis process is also one of the plasma ammonia synthesis technologies [229].The device is comprised of a cylindrical quartz tube,where the plasma is generated by an RF generator in a matched network of 120 W in the tube,in the presence of a catalytic surface rolled foil(here tungsten purity is 99.97%),on the other side of the quartz tube is the residual gas analyzer (RGA) which serves to analyze the gas species generated by the plasma(Fig.29(c)).In the plasma phase,NHxradicals are generated by the reaction between dissociated atoms and excited molecules that can produce ammonia.When a tungsten catalyst was introduced,the nitrogen preloaded on the W surface was exploited to form ammonia in the hydrogen plasma,and the amount of ammonia increased following the addition of nitrogen.Impressive ammonia yields of 32%were obtained at 25/75% N2/H2.The results of the RGA study indicated that the maximum yield of ammonia on the W surface was obtained at the same initial concentration of nitrogen and hydrogen,An Eleyriddel mechanism pathway was proposed giving for ammonia formation in this process.It was demonstrated that zeolite imidazolate frameworks (ZIFs) were effective catalysts for non-thermal plasma ammonia and the experiments were carried out in a(DBD)reactor [228].The internal electrode was made of tungsten wire and was placed on a quartz tube.The external electrode was made of tin-copper mesh.The catalysts were filled in overlap zone between the internal and external electrodes.The ammonia synthesis rate of ZIF-67 and ZIF-8 was 26.65 and 28.52 μmol∙g-1-∙min-1,respectively (Fig.29(d)),exceeding the other catalysts under the same reaction conditions.The active sites of the ZIFs were organic linkers of —C=C-,which was a polarizable group.There is no straight relationship between hole dimensions and performance of catalysis.The weaker storage capacity was generated due to feeble interactions of dipole–dipole and polar walls of ZIF,resulting in more escaped ammonia production.Also,the presence of metals(Zn and Co,respectively)might promote higher ammonia synthesis rate.The presence of argon produced a great rate which was 1.2 folds higher than without argon owing to the improvement of plasma homogeneity.In addition,the low specific energy input(SEI) values caused by argon indicated that Ar increased the concentrations of nitrogen vibrational species,and the values of N2+plasma excitation species were higher than those of atomic nitrogen,requiring less energy than atomic nitrogen.
CoNPs with variable sizes might be obtained by extending the distance between Co atoms on the addition of Zn2+that could substitute a portion of Co2+sites,showing extraordinary activity.And the optimal catalyst was Co-N2(Fig.30(a)) [230].The optimal plasma-catalyzed catalysts for ammonia synthesis were explored from Co monoatomic catalysts to Co nanoparticles with diverse sizes and various Co-N coordination numbers (Fig.30(b)).The nitrogen yield varied with Co particle size,as the smaller size of the Co NPs,the greater the number of exposed active centers,which further increased the catalytic efficiency.N2was readily activated by adsorption-dissociation on the surface of Co-N2catalysts,which was also demonstrated by theoretical calculations.The N2reduction mechanism of SAC was inclined to follow the alternating pathway.Chalcogenides were capable of spontaneous and permanent electrolysis in the existence of an external electric field.MgTiO3and other chalcogenides,which acted as potent catalysts for chilled plasma synthesis and integration of ammonia,were studied[231].MgTiO3chalcogenides had the fastest ammonia synthesis rate of 12.16 μmol∙m-2∙min-1.The high electronegativity of Mg could facilitate the dissociation of the tri-nitrogen covalent bond.The dielectric constant value of this chalcogenide could improve the homogeneity of the plasma.The ammonia production capacity of MgTiO3chalcogenide was almost twice as much as that of conventional oxides and some micellar crystals(Fig.30(c)).They also discussed that the superiority of MgTiO3chalcogenide over others could be attributed to its high dielectric constant,large electronegativity,and functional porous crystal structure.The addition of He resulted in the increase of ammonia yield.At higher power,there was the lowest energy yield attributed to the ammonia decomposition reactions,and the efficiency increased with the increase of plasma power.Therefore,it was necessary to keep the reaction at low power and separate the synthesized ammonia in time.The highest ammonia decomposition rate of 44.37% was obtained at 20 W (Fig.30(d)).
Fig.30. (a)Sketch of the Co-N2 coordination number catalyst reaction.(b)Influence of varying nitrogen coordination numbers of CoSACs on ammonia synthesis.Reproduced from Ref.[230]with permission of American Chemical Society,copyright 2021.(c)Reflection of ammonia production rates of different alkaline earth metal chalcocite under 1:1 condition.(d) Ammonia synthesis rate of MgTiO3 at variable power under differential atmosphere.Reproduced from Ref.[231] with permission of Royal Society of Chemistry,copyright 2021.(e)Non-thermal plasma-electrochemical ammonia synthesis process.(f)Schematic diagram of a small-scale plasma combined within an electrical system.(g) Amine production rate and Faradaic efficiency as a function of voltage.(h) Ammonia equivalent yield and energy consumption of plasma electrochemical combination systems and other NRR systems.Reproduced from Ref.[232]with permission of Royal Society of Chemistry,copyright 2021.(i)PNOCRA process with two stages:plasma-assisted N2 oxidative adsorption;NOx reductive extraction to ammonia.Reproduced from Ref.[233] with permission of Wiley Online Library,copyright 2020.
Plasma modification induced by catalysts may also enable the reactions on or near the catalyst surface to be modulated by local changes in the electric field.The dielectric materials are commonly positioned in the discharge region of the plasma,The metal catalyst carrier will modify the behavior of the plasma by affecting the electric field intensification and the electron thermoregulation,which will change the plasma activation [12].There are relatively few studies on this aspect: the differential plasma synthesis of ammonia on Ni/Al2O3,Co/Al2O3and Fe/Al2O3with DBD have been explored.Despite discrepancies in ammonia production rates of diverse materials,there is no apparent differences across plasma currents,filament counts or capacitances.Hence,the disparity of ammonia production rates is linked to inherent catalytic activity of catalyst rather than alterations in the plasma,however,the indepth mechanism remains to be investigated.
Unlike the aforementioned catalyst-plasma interaction,Sunet al.[232] has developed a continuous approach to produce ammonia which has combined plasma and electrochemical technology,the air was activated by non-thermal plasma to produce NOx and then converts the synthesized NOxto ammonium electrochemically(Fig.30(e),(f)).The primary reaction was undertaken in a plasma bubble column reactor,where a glow discharge system(prone to nitrate production) and a spark discharge (prone to NOxproduction)were assembled to leverage the advantage of each potential,the interaction between the plasma and the liquid benefited the generation of hydroxyl radicals and transport of the generated reactive material (NOx).CuNWs were able to achieve the greatest plasma current density which could reduce plasma activation of the electrolyte and attain ammonium production rate of 45 nmol∙cm-2∙s-1with FE close to 100%.The ammonium production rate (NH) and FE grew as the potential varied from 0.2 to–0.6 V (Fig.30(g)).At lower potentials NOwas converted to NOwhich was generated by dissolving*NO2in water.The conversion of NOto ammonia could be induced at high potentials.As presented in Fig.30(h),this approach had a better overall rate of ammonia production than the other three recently reported approaches.The advantages of well-scalable potential and low energy input might contribute to a leap towards large-scale chemical synthesis in plasma.Hollevoetet al.[233]initiated the plasma nitrogen oxidation and catalytic reduction to ammonia (PNOCRA)process,also a merger of plasma-assisted nitrogen oxidation and electrochemical technology (Fig.30(i)).PNOCRA consumed less energy and meet the requirement of 4.6 MJ∙mol-1.The gaseous mixture was fed into a plasma reactor with operating temperature of 1100 °C for conversion to compounds such as NO.The exiting plasma reactor gas traveled through a heat exchanger operating at 175°C and was consequently adsorbed and converted to ammonia on a lean NOxtrap,which was chilled to 40 °C enabling the extraction of ammonia with liquid water accompanied by the recycling of H2.The depleted NOxtrap has a double function,i.e.,the adsorbed NOxmay be convertible to N2or NH3under O2conditions,relying on the selectivity of catalyst(Ba(NO3)2+8H2→2NH3+BaO+5H2O;Ba(NO3)2+5H2→N2+BaO+5H2O).The accelerator in this case was Pt/BaO/Al2O3,which was embedded on a cordierite honeycomb monomer.Considering the different reaction rates of two units,the amount of simultaneous work should be modulated to prevent gas clogging and also avoid frequent re-starting of the plasma reactor.The engineered process comprised three energy consuming units: the plasma reformer,the electrolytic cell and the ammonia splitter,of which the plasma reformer is the most expensive.Muzammiet al.[234] presented a novel,sequential,expandable,mono-step plasma catalyst assembly technology recently,that advanced the development of sustainable and environmentally friendly ammonia production.N2was inputted into the reactor through two holes in the bottom of a cylindrical floor,droving a rotating arc.Water film further produced H and OH/O radicals by reacting with nitrogen,subsequently to form NOxwhich was in the Pd/γ-Al2O3catalyst system to produce a high ratio of NH3(95%) at a relatively fast rate (120 μmol∙s-1).
7.Summary and Outlook
The methods of ammonia synthesis have been summarized and their properties and development trends are highlighted in Table 9.The Haber Bosh process is as an unprecedented and pioneering achievement in the history of ammonia synthesis,catalyst with low energy consumption and high efficiency is at the most fundamental core of the process.In the field of thermocatalytic ammonia synthesis,Ru based catalysts and Fe based catalysts have been well and extensively investigated,leading to the kellogg advanced ammonia synthesis process.Ammonia synthesis catalyst such as metal composites and metal-free materials have been investigated in 21st century,however due to the heavy use of fossil fuels,the exploration of thermocatalysts is not a dominant aspect of contemporary ammonia research.The derivative chemistry looping has also been vigorously investigated,particularly the aspects of the thermodynamics of nitrogen fixation and diverse nitrogen carriers,and the low efficiency of ammonia yield is ultimately the main impediment for the application of this process.For H2O-CL,the conversion of metal oxides to metals or straightforward nitrogenation is frequently thermodynamically disadvantageous,which hashigh kinetic resistance and demands harsh operating conditions.It is worthwhile to investigate and evaluate the feasibility of developing ternary or multiple oxides to lower the thermodynamic stability of binary metal oxides.Compared to the H2O-CL process,the TM nitrides such as manganese nitride are moderately thermodynamic and have high kinetic resistance and low ammonia production.Alkali (earth) metal imides,owing to moderate thermodynamics of nitrogen fixation and hydrogenation,have exhibited excellent low temperature kinetics and highest ammonia yield rate involving chemical looping.During the 21st century,facing the challenges of the transformation of the modern economy and the increasing pollution of the environment,ammonia,having advantage of easy transport and storage,is deemed to be the desired energy carrier that might cope with the energy crisis,electrocatalysis and photocatalysis with carbon emissions are considered as two alternatives to the high pollution thermos-catalytic technology.It is possible to synthesis NH3by utilizing plentiful N2and H2O as feedstock under mild conditions.It is possible to store clean and renewable solar energy and electricity in the form of hydrogen carriers and carbon-free fuels(NH3).The NRR activity of the Au and Ru-based noble metals catalysts is dramatically affected by the composition,particle dimensions and support.Non-noble metal-based materials such as Bi-based,Mo-based and other based metal-free material classes can achieve competitive NRR activity by modification of surface alignment,structure and electronic properties.Carbon-based materials,Bi and S-based materials,have been extensively investigated for their unique properties and structures,achieving good efficiencies and considerable path to application.Despite of significant developments in the electrochemical production of ammonia,it is more suitable for practical applications to the Haber process after the electrolytic production of hydrogen.Firstly,because the Haber process is now well established and the development of catalysts has led to significant promotion in pressure and temperature conditions.Secondly,the hydrogen produced from the electrolysis of water is not only used for ammonia production,but also contributes to solving other energy problems.Likewise,we have summarized recent achievements in photocatalytic ammonia synthesis,particularly photocatalysts and design strategies,pointing the direction for improvements of photocatalysts for N2activation and reduction.The premier work in light-driven NRR has concentrated on the research of titanium oxide-based catalyst,including approaches of interfacial manipulation,surface engineering,and chemical modification.The pivotal contribution of oxygen vacancies in photocatalyst systems is that it arouses the investigational interest in defect engineering.Crude surfaces rich in defects and doped heteroatoms have a greater catalytically active surface area and a considerable number of low-coordination sites,which facilitates an increase in the rate and selectivity of ammonia synthesis.The fabrication of heterostructure and modifications of other photocatalyst are currently being explored on a large scale (e.g.,porous metal–organic frameworks,nanoclusters,black phosphorus) in order to enhance optical utilization and inhibit HER.In summary,a more accurate and systematical design of catalysts is necessary to achieve a breakthrough in activity and selectivity.It has been theoretically proposed that the circumvention of scale relationships may lead to a leap in electrical/optical NRR performance.However,specific methods of achieving this goal remain to be implemented.

Table9 Five types of ammonia production characteristics and development directions
In addition to exploitation of catalyst,for photochemical production,light collectors,reaction systems and reaction conditions are also indispensable on the path to industrialization,like electrode structure design,electrolyte composition and non-aqueous system construction for electrochemical production.The optimization of systems,exacting methods for ammonia quantification and means of assessing catalytic performance should keep up to date with further enhancements.The three main methods for testing ammonia,Nessler’s reagent colorimetric method,the indophenol blue method and the salicylate method,together with various electrolytes,are also essential components of an electro-ammonia system.However,because of the considerable complexity of the reaction,we still have a very ambiguous understanding of the mechanism of photo/electric nitrogen fixation,while progress in heterogeneous catalytic nitrogen fixation is facing major roadblocks,such as: the difficulty of activation of N≡N triple bond,the weak absorption of N2on the heterogeneous catalyst,the multi-electron and proton participation pathways.In general,the manufacture of ammonia accompanied by the generation of hydrogen and hydrazine as side reactions,resulting in unsatisfactory FE.The overall energy usage for technical ammonia synthesis is 3.5×104–5×104J∙g-1.If the FE of an electrocatalytic system were to exceed 50%,as a result,the total overall energy consumption would be approximately 1.9 × 105J∙g-1.For photocatalytic systems,a 10% conversion efficiency would result in 2.1 × 105J∙g-1[235].Thus,it is only by satisfying the above energy requirements that a path towards industrialization can be opened.
In order to overcome the shortcomings of photovoltaic/electric ammonia production,we have summarized the vane of ammonia development as follows:Firstly,continue to develop low-cost,efficient multi-species catalysts.Quantum dots,nanowires,singleatom SACs and other catalysts with various compositions and morphologies have emerged as recent trends in research.Secondly,it is also imperative to unravel the fuzzy underlying mechanisms.The effective combination of theoretical analysis and experimental can be beneficial in revealing the reaction mechanism of NRR at the molecular scale.Thirdly,it is recommendable to adopt advanced characterization techniques to explore the accurate active site and structural properties of intermediates by shifting from conventional characterization methods such as single SEM,TEM and XPS to aberration corrected transmission electron microscope,diffuse reflectance Fourier transform infrared spectroscopy and EPR.In addition,isotopic labelling combining with Fouriertransform infrared spectroscopy and mass spectrometry monitoring is an obvious and straightforward way to detect the source and route of reaction products.On-line ammonia detection methods are designed to obviate the use of spectrophotometric determination which is time-consuming and inaccurate.This paper also complements the plasma intervention-assisted ammonia synthesis method.The plasma approach is implemented by subdividing the nitrogen fixation process.It is a new subspecies of ammonia production obtained by the combination of two parts.The gas is inserted into the plasma to achieve the production of intermediates such as NOxin the former part and the intermediates are experimented through thermal or electrocatalytic processes in the later part.The different discharge methods of the sliding arc,plasma jet and dielectric barrier discharge,as well as the type,structure and characteristics of catalyst,all affect the effectiveness and applications of the NRR.
The present ammonia generation method is still based on Haber process.Although milder synthesis methods have achieved many impressive results in the laboratory,there are a host of tricky reasons such as costly equipment,environment and transition that have hindered their industrialization,as shown in Table 9.Among them,although the electro/photocatalytic ammonia technology has one shortcoming or another,a variety of systems have emerged that utilize light and electrical energy as power in combination with the Haber method.Furthermore,miniaturized photo/electrocatalytic ammonia plants are expected to achieve‘distributed’‘ondemand’ ammonia production,complementing the existing large,centralized large-scale chemical plants.The future electrocatalytic ammonia production may be prioritised for industrialization in view of solar and electrical energy development trends.Hence the exploration of catalysts and processes that are less energy intensive,more conducive to industrial transition and more cost effective,remains the focus of investigation.
CRediT Authorship Contribution Statement
Supeng Yu:Writing– original draft.Ting Xiang:Writing–review &editing.Njud S.Alharbi:Writing– review &editing.Bothaina A.Al-aidaroos:Writing– review &editing.Changlun Chen:Writing– review &editing,Supervision,Project administration.
Data Availability
No data was used for the research described in the article.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Financial supports from the National Natural Science Foundation of China (22276194),Institute of Energy of Hefei comprehensive National Science Center (21KZZ501 and 21KZS201),and the Presidential Foundation of Hefei Institutes of Physical Science,Chinese Academy of Sciences (YZJJZX202019).This work was funded by the Deanship of Scientific Research (DSR),King Abdulaziz University,Jeddah,Saudi Arabia under grant (KEP-PhD: 65-247-1443).The authors,therefore,acknowledge with thanks DSR technical and financial support.
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cjche.2023.03.028.
 Chinese Journal of Chemical Engineering2023年10期
Chinese Journal of Chemical Engineering2023年10期
- Chinese Journal of Chemical Engineering的其它文章
- High catalytic performance of CuCe/Ti for CO oxidation and the role of TiO2
- Experimental and numerical studies of Ca(OH)2/CaO dehydration process in a fixed-bed reactor for thermochemical energy storage
- Volumetric and ultrasonic properties of thiamine hydrochloride drug in aqueous solutions of choline-based deep eutectic solvents at different temperatures
- Synthesis of zeolite A and zeolite X from electrolytic manganese residue,its characterization and performance for the removal of Cd2+ from wastewater
- Metal-organic framework-derived Co-C catalyst for the selective hydrogenation of cinnamaldehyde to cinnamic alcohol
- A pseudo transient nonequilibrium method for rigorous simulation of multicomponent separation columns