
Metal-organic framework-derived Co-C catalyst for the selective hydrogenation of cinnamaldehyde to cinnamic alcohol
2023-12-31 04:02FupingTianXinchiZhangYingyingShengXiaoChenXiangWangChanghaiLiang
Fuping Tian,Xinchi Zhang ,Yingying Sheng ,Xiao Chen ,Xiang Wang ,Changhai Liang
1 State Key Laboratory of Fine Chemicals &Department of Chemistry, Dalian University of Technology, Dalian 116024, China
2 School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China
Keywords:Catalyst Hydrogenation Selectivity Cinnamaldehyde Cinnamyl alcohol Co-C
ABSTRACT The liquid phase selective hydrogenation of cinnamaldehyde has been investigated over the catalysts Co-C-T(T=400–700°C),which were derived from the carbonization of the MOF precursor Co-BTC at different temperatures in inert atmosphere.Co-C-500 exhibited a higher conversion (85.3%) than those carbonized at other temperatures,with 51.5% selectivity to cinnamyl alcohol,under a mild condition(90 °C,4 h,2 MPa H2,solvent: 9 ml ethanol and 1 ml water).The high catalytic activity of Co-C-500 can be ascribed to the large specific surface area of the catalyst,the uniformly dispersed metallic cobalt nanoparticles,and the more defect sites on the carbon support.Moreover,Co-C-500 showed excellent reusability in 5 successive cycles,mainly related to the uniformly dispersed cobalt nanoparticles embedded in carbon support.
1.Introduction
The selective hydrogenation of α,β-unsaturated aldehydes has received lots of attention because their products are widely used in fine chemical industries [1].Cinnamaldehyde (CAL) is one of the representative reactants,and its selective hydrogenation reaction is often used as a probe reaction to analyze the hydrogenation mechanism of C=C and C=O.The main products include cinnamyl alcohol (COL),hydrogenated cinnamaldehyde (HCAL) and hydrogenated cinnamyl alcohol (HCOL) (Fig.1).COL is a product of the hydrogenation of C=O bond with high application value,which is often used as fragrance,flavor,food additive and pharmaceutical intermediate [2].However,from the viewpoint of thermodynamics,hydrogenation of C=C is more favorable than C=O,since the bond energy of C=C (615 kJ∙mol-1) is less than that of C=O(715 kJ∙mol-1) [3].Moreover,the conjugated structure between C=O and C=C groups(Fig.1)brings about the simultaneous hydrogenation of them,generally resulting in a mixture of three hydrogenated products.Therefore,the selective hydrogenation of CAL to COL is still one of the important challenges in current research.
In the heterogeneous catalytic hydrogenation of CAL,noble metal-based catalysts have been extensively studied,such as Pd-,Pt-and Ir-based catalysts [4–6].They exhibit high hydrogenation activity,in which Pt-and Ir-based ones tend to catalyze C=O hydrogenation.However,the high price of precious metals due to the scarcity limits their practical application.Compared with precious metals,non-precious metals are more abundant and affordable.Therefore,there is a great interest in studying non-precious metals,including Co,Ni and Cu,as substitutes for precious metal catalysts.It has been reported that the catalytic activity and selectivity of Cu-based catalysts for CAL hydrogenation were not satisfactory [7,8].For instance,Dragoiet al.[7] prepared Cu/SBA-15 catalyst by the impregnation method,even the optimal catalyst Cu10/SBA-15 achieved 40%CAL conversion(59.6%HCAL selectivity)at 130 °C,1 MPa H2.Comparatively speaking,the Ni-based catalysts displayed moderate activity and preferred hydrogenation of C=C bond [9–11].For example,Liet al.[11] synthesized spherical sandwich-like catalyst mSiO2@Ni/SiO2@mSiO2,which exhibited excellent catalytic performance under mild conditions (100% conversion of CAL with 100% selectivity to HCAL at 100 °C and 3 MPa H2for 40 min),though the preparation method was a little bit tedious.

Fig.1. Reaction routes of CAL hydrogenation.
Among the extensively investigated non-precious metal catalysts,only Co-based catalysts showed moderate activity and exhibited a tendency to C=O hydrogenation [12].For example,Bustamanteet al.[13] prepared Co-CoO@SiO2core–shell catalysts by reduction under H2atmosphere,and the modulation of active components was achieved by controlling the reduction temperature.The optimal catalyst displayed 68% selectivity to C=O hydrogenation,but 60.0% conversion of CAL was unsatisfied at 70 °C,4 MPa H2for 4 h.To further improve the catalytic performance,some strategies,such as introducing a second metal component or doping heteroatoms,were usually employed by researchers [14–17].For example,Jianget al.[18] loaded cobalt nanoparticles onto nitrogen-doped porous carbon materials(CPNs)viaone-pot method.Compared to the N-free catalyst Co@C,the N-doped catalyst Co@CPNs achieved a high selectivity to hydrogenation of C=O (80%) with complete conversion of CAL (99%).However,a somewhat harsh reaction condition (temperature 180 °C,3 MPa H2) was required.Ciotoneaet al.[19] reported an increased catalytic activity of Co/SBA-15 by introducing the second metal Cu.The optimal catalyst CuCo1:4/SBA-15 showed 99% conversion of CAL and 72% selectivity to COL at 24 h (temperature 150 °C,atmospheric hydrogen),while the control catalyst Co/SBA-15 only achieved 55% conversion of CAL.The authors attributed the improved catalytic performance to a synergistic effect between Co and Cu.So far,except for Co,there are few reports that non precious metals were used as catalysts alone for CAL hydrogenation to COL.Moreover,the research reports on Cobased catalysts are quite limited in the literature.These Co-based catalysts generally required higher temperatures,larger hydrogen pressure,or prolonged reaction time to achieve high catalytic performance [18].Therefore,it is still a challenge to develop cobaltbased catalysts with high catalytic performanceviasimple and efficient preparation methods.
Carbon materials are widely used as the carriers to support the metal/metal oxide nanoparticles in chemical,energy and environmental fields due to the high specific surface area and porous structure [20,21].In recent years,metal organic frameworks(MOFs)consisting of inorganic metal ions and organic ligands have been widely used as sacrificial templates to obtain porous carbonsupported metal/metal oxides through thermal decomposition in inert atmosphere.The derived materials to a large extent retain the characteristics of the MOFs precursors,such as regular morphology,high specific surface area,and rich pore structure.As a consequence,the derived carbon-supported metal materials exhibit excellent thermal stability and better dispersion of active metal[22,23],and therefore have been widely studied in various catalytic reactions,such as oxidation [24,25],hydrogenation [26-28] and Fischer-Tropsch reactions [29].In the hydrogenation reaction of unsaturated aldehydes and ketones,Zhanget al.[30] prepared Co@C catalysts by carbonizing Co-MOF-74 precursor at 600 °C for 2 h under Ar atmosphere and used it as the catalyst for the hydrogenation of CAL,giving 40.1%conversion of CAL and 61.1%selectivity to COL at 60 °C under 1 MPa H2for 8 h.Though the carbon-coated structure of this catalyst played a positive role in stabilizing the active species,the exposed active sites after carbonization were not enough due to the structural characteristics of the precursor,resulting in an unsatisfied catalytic activity.In addition,Suet al.[31] reported the preparation of carbon-coated Ni (Ni/C) catalysts by the pyrolysis of a sacrificial template Ni-MOFs (Ni-BTC),and the resultant Ni/C catalyst showed good catalytic activity in the selective hydrogenation of furfural.The optimal catalyst Ni/C-500 displayed 100% conversion of FAL and 100% selectivity to THFOL at 120 °C under 1 MPa H2for 2 h,ascribed to the better dispersion of Ni metal and a high specific surface area (92 m2∙g-1).The above studies demonstrates that the preparation of porous carbon-coated metal/metal oxide catalysts using MOFs as precursors is a promising synthetic method.
In this work,Co-BTC was synthesized as a precursor by a simple solvothermal method,and then it was carbonized at different temperatures under Ar atmosphere to prepare Co-C-Ts catalysts for the selective hydrogenation of CAL.The decomposition of organic ligands in the precursors facilitates the formation of porous structures in the carbon support and uniform dispersion of Co species in the derived catalysts.The experimental results showed that the optimal catalyst Co-C-500 reached 85.3% of CAL conversion and 51.5%of COL selectivity at 90°C under 2 MPa H2for 4 h.The facile preparation,high activity,good stability,and easy recovery of Co-C-500 make it a promising catalyst in hydrogenation reaction.
2.Experimental
2.1.Materials
The reagents used in this experiment are all analytical pure and directly used without any purification.1,3,5-tribenzoic acid(H3BTC),cinnamaldehyde(CAL),cinnamyl alcohol(COL),hydrocinnamaldehyde (HCAL),and hydrocinnamyl alcohol (HCOL) were purchased from Aladdin reagent Co.,Ltd,China.Co(NO3)2∙6H2O,isopropanol (IPA),tetrahydrofuran (THF),and toluene were purchased from Tianjin Damao chemical reagent Co.,Ltd,China.Methanol (CH3OH),ethanol (EtOH),andN,N-dimethylformamide(DMF) were purchased from Xilong Scientific Co.,Ltd,China.
2.2.Preparation of catalysts
2.2.1.Preparation of Co-BTC
Typically,Co(NO3)2∙6H2O(2.44 g,8.4 mmol)and H3BTC(1.00 g,4.8 mmol)were dissolved in 51 ml of the solution containing DMF,ethanol,and deionized water (volume ratio=1:1:1).After sonication for 30 min,a uniform and transparent violet solution was obtained,transferred to several glass vials,sealed,and crystallized in oven at 85 °C for 20 h.Then it was cooled to room temperature(RT),and the acicular solid product was separated by filtration and washed with H2O and ethanol for several times to obtain Co-BTC.
2.2.2.Preparation of Co-C-T
At first,550 mg Co-BTC was dried in an oven at 120°C for 12 h.Then,the dried Co-BTC was placed in a quartz boat,carbonized in a tubular furnace under Ar atmosphere (100 ml∙min-1).The sample was heated from RT to the desired temperatures(400,500,600 and 700 °C) at 5 °C∙min-1and kept at that temperature for 2 h.The resulting samples were cooled in Ar atmosphere and labeled as Co-C-T,whereTdenotes the carbonation temperature.The whole procedure was illustrated in Fig.2.

Fig.2. A scheme of Co-C-T preparation.
2.3.Characterization
X-ray diffraction (XRD) patterns were taken on a SmartLab(Japan) 9 KW X-ray diffractometer using Cu-Kα (λ=0.15418 nm)at 45 kV and 100 mA and collected in the 2θ range of 5°–50° for the MOF precursor or 5°–80°for Co-C-Tcatalysts.Thermogravimetric analysis(TGA)of Co-BTC(10 mg)was performed on a TGAQ500(TA Instruments)by heating the precursor MOF from 30°C to 800°C at a ramp rate of 10°C∙min-1in N2flow(60 ml∙min-1).Cobalt contents of the samples were tested by inductively coupled plasma atomic emission spectrometry (ICP-AES) on PerkinElmer Nex ION 300D.The skeletal structure of the samples was determined by the KBr pellet method on Nicolet Avatar 360 Fourier-Transform Infrared spectrometer (FT-IR) by scans of 64 with a resolution of 4.0 cm-1in the 4000–400 cm-1range.An inVia Qontor Raman Microscope(Renishaw,UK)was used to analyze the surface defect sites of the samples.The samples were scanned over a range of 50–4000 cm-1with an excitation light wavelength of 532 nm.The N2adsorption–desorption isotherms of Co-C-Twere determined on the Micromeritics ASAP 2020 Plus at-196°C.About 150 mg of the sample was degassed at 200°C for 5 h prior to testing.The scanning electron microscopy(SEM)measurements were performed on a Quanta 450 instrument with an accelerating voltage of 20 kV.The transmission electron microscope (TEM) images were obtained at 200 kV with a JEM-F200 manufactured by Nippon Electron Co.,Ltd.An Escalab Xi+X-ray photoelectron spectrometer (XPS) from Thermo-VG Scientific was used for the analysis of the surface composition of the Co-C-Tsamples.A monochromatic Al Kα X-ray excitation source(1486.6 eV)was used.The software XPS PEAK-41 was employed to fit the obtained data.
2.4.Catalytic test
The catalytic performance of the catalysts Co-C-Twas evaluated by using CAL hydrogenation as the probe reaction.The reaction was conducted in a 25 ml stainless autoclave,which was loaded with 9 ml ethanol,1 ml H2O,1.5 mmol CAL,and 20 mg catalyst.The autoclave was sealed and the air in it was fully replaced with 2 MPa H2for 3 times.After that,the autoclave was charged with hydrogen (1.0–3.0 MPa) at RT.The stirring speed was set at 900 r∙min-1to eliminate the diffusion effect.At the end of the reaction,the reactor was rapidly cooled down to RT in ice-water bath.The reaction solution was quantitatively analyzed by GC-7890F (Tianmei) and FID detector with FFAP column (30 m × 0.32 mm × 0.5 μm),using relative response factor method.The temperatures of the column,the detector and the inlet are set at 190 °C,280 °C,and 280°C,respectively.In the recycling runs,the catalyst attached to the magnetic stirrer was washed with ethanol for 4 or 5 times after the reaction,and then was directly put into the reaction solution for the next run.
3.Results and Discussion
3.1.Characterization of the MOF precursor
The XRD pattern of Co-BTC precursor is presented in Fig.3(a).As can be seen,the position of all diffraction peaks of the assynthesized Co-BTC is consistent with that of the simulated one(CCDC 1274034),indicating that the Co-BTC precursor has been successfully synthesized[32].The intensity of the diffraction peaks is quite different from the simulation results,which is related to the different preferred growth planes of the materials.TG analysis was carried out in N2atmosphere to determine the carbonization temperature range of Co-BTC,and the result is shown in Fig.3(b).Two obvious stages of mass loss can be observed during the heating decomposition of Co-BTC.The first stage occurred in the temperature range from 100 °C to 200 °C by a mass loss rate of 25.8%,which can be attributed to the volatilization of residual solvent and lattice water.In the second stage,the mass decreased sharply (36.4%) in the temperature range of 400–550 °C,which can be attributed to the decomposition of Co-BTC skeleton.When the temperature was further raised above 600 °C,the mass of the residue remained basically stable [31,33].Based on the above results,the decomposition of Co-BTC started at 400 °C,happened vigorously between 450–550 °C,and almost completed at 600 °C.Therefore,the precursor Co-BTC was carbonized at four different temperatures (400,500,600,700 °C),and the derived sample was denoted Co-C-T,whereTstands for carbonization temperature.
3.2.Characterization of Co-C-Ts
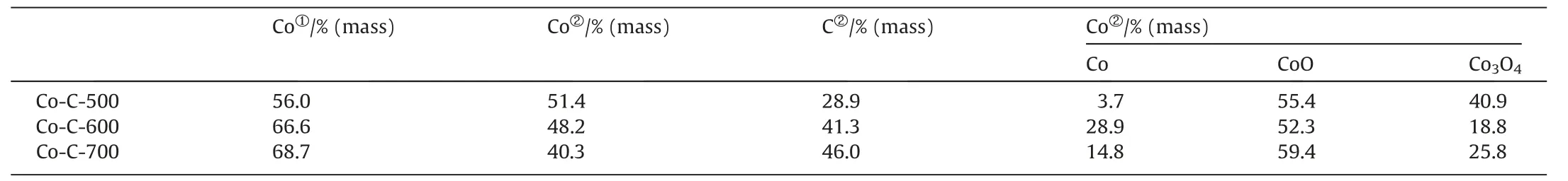
The FT-IR spectra of the Co-C-Tcatalysts were collected in the range of 4000–500 cm-1,and the results are shown in Fig.4(a).The precursor Co-BTC was included for comparison.For Co-BTC,the broad peak around 3700–3100 cm-1can be attributed to the stretching vibration of O-H,indicating the presence of crystalline water in the synthesized MOF material.The peaks at 1560 cm-1and 1376 cm-1can be designated to the symmetric and asymmetric stretching vibration of carboxylate group,respectively.The characteristic vibration at 723 cm-1might be attributed to Co-O stretching vibration,arising from the coordination of the carboxylate oxygen atom in BTC ligand with Co [34].However,no characteristic vibration peak of C-OH was observed near 1720–1680 and 1275 cm-1,suggesting that H3BTC deprotonated and complexed with metal ions to form Co-BTC precursor [35].For Co-C-T,the absorption peak intensity of the above functional groups decreased with the increase of carbonization temperature.It should be noted that the positions of the absorption peaks of Co-C-400 were approximately the same as those of the precursor,but the intensity of the peaks became significantly lower,indicating that the structure of the precursor retained to a large extent and the carbonization was not yet completed for Co-C-400.The above absorption peaks basically disappeared when the carbonization temperature reached or exceeded 500 °C,indicating that the organic framework of Co-BTC collapsed and the carbonization was completed.The information derived from the FT-IR test is generally in accordance with the TG result.In addition,the Co contents in Co-C-400,Co-C-500,Co-C-600 and Co-C-700 samples were determined to be 32.4%,56.0%,66.6% and 68.7% (mass) respectively by ICP-AES.The lower Co content in Co-C-400 confirmed again that the organic ligand didn’t completely decompose at 400°C.Meanwhile,considering the fact that Co-C-400 showed no catalytic activity in CAL hydrogenation,no further characterization is performed on Co-C-400.

Fig.3. (a) XRD patterns and (b) TG (DTG) curves of Co-BTC.

Fig.4. (a) FT-IR spectra of Co-BTC and Co-C-T;(b) XRD patterns,(c) Raman spectra and (d) N2 adsorption–desorption isotherms of Co-C-T at different carbonation temperatures.
The XRD patterns of Co-C-Tcatalysts are shown in Fig.4(b).As can be seen,three diffraction peaks were observed at 2θ=44.2°,51.5°,and 75.9°,corresponding to(1 1 1),(2 0 0),and(2 2 0)crystal planes of metal Co (JCPDF-#15-0806),and the other three diffraction peaks at 2θ=36.5°,42.4° and 61.5° can be designated to(1 1 1),(2 0 0),and (2 2 0) crystal planes of CoO (JCPDF-#43–1004).In addition,two weak diffraction peaks can be observed on Co-C-500 at 2θ=36.9° and 44.8°,which are assigned to the(3 3 1) and (4 0 0) crystal plane of Co3O4(JCPDF-#42-1467).In Co-C-600 and Co-C-700,the diffraction peaks ascribed to Co(0)became narrow and the intensity increased,implying the agglomeration of Co nanoparticles with the increased carbonization temperatures.
Raman spectra were employed to analyze the state of the carbon matrix in the catalysts Co-C-T.As displayed in Fig.4(c),two characteristic Raman peaks at 1335 cm-1and 1596 cm-1were observed on three samples,representing D band and G band,respectively [36].According to the literature,the strength ratio of D band to G band (ID/IG) is directly proportional to the amount of structural defect sites in the carbon matrix and inversely proportional to the degree of graphitization [37].TheID/IGvalues of Co-C-500,Co-C-600 and Co-C-700 decreased from 2.47 to 1.66 as the carbonization temperature increased.It has been reported in the literatures that the defect sites on the carbon matrix affected the catalytic performance.On one hand,a large number of unpaired π-electrons existing in the carbon matrix can effectively accelerate the electron transfer and reduce the formation energy of key intermediates[38].So,the defect sites in the carbon matrix in Co-C-500 may contribute to the acceleration of electron transfer and therefore the catalyst displays high catalytic activity in CAL hydrogenation.On the other hand,more defect sites in the carbon matrix can facilitate the anchoring of the metal nanoparticles and promote their uniform dispersion [39].Therefore,Co-C-500 with more defective sites on the carbon matrix exhibited higher catalytic activity on CAL hydrogenation than the other two catalysts.
The textural properties of Co-C-Twere measured by N2adsorption–desorption isotherms (shown in Fig.4(d)),and the data are listed in Table 1.As can be seen,the adsorption and desorption isotherms of three catalysts are similar,and the hysteresis loops can be clearly observed,meanwhile,certain amount of adsorption volume was observed in the low specific pressure region,indicating the presence of both microporous and mesoporous structures in the samples.The total pore volume and BET surface area of Co-C-500 were determined to be 0.12 cm3∙g-1and 223 m2∙g-1,respectively,higher than those of Co-C-600 (0.10 cm3∙g-1and 171 m2∙g-1)and Co-C-700(0.10 cm3∙g-1and 190 m2∙g-1)(Table 1).The N2adsorption–desorption results suggest that the carbonization temperature of 500°C was beneficial to the formation of abundant pore structure,due to the decomposition and condensation of the organic skeleton of the precursor;when the carbonization temperature was further increased to 600°C or 700°C,further condensation of the organic skeleton or the agglomeration of Co NPs may lead to the decrease of specific surface area and pore volume.
SEM and TEM are used to characterize the microscopic morphologies of the samples.The SEM images are shown in Fig.5(a)–(d).The Co-C-Tcatalysts are block-like as a whole,with layered structures inside and rough surface.The TEM images are displayed in Fig.5(f)–(h),the metal particles in Co-C-Tare uniformly distributed in the carbon support.It is found that the average particle sizes of Co NPs increase slightly from 11.3 nm for Co-C-500 to 12.8 nm for Co-C-600 and 14.7 nm for Co-C-700,indicating that the agglomeration of Co NPs occurred as the carbonization temperature increased.In addition,the high-resolution transmission electron microscopy(HR-TEM)image of Co-C-500 is shown in Fig.5(e).A lattice fringe of 0.205 nm ascribed to the Co(1 1 1)crystal plane can be clearly observed.The inset in Fig.5(e)shows the SAED pattern of Co-C-500,only a weaker diffraction ring of Co(1 1 1)can be observed,implying the lower crystallinity of metallic Co in Co-C-500,which also agrees with the wide diffraction peaks in the XRD results.
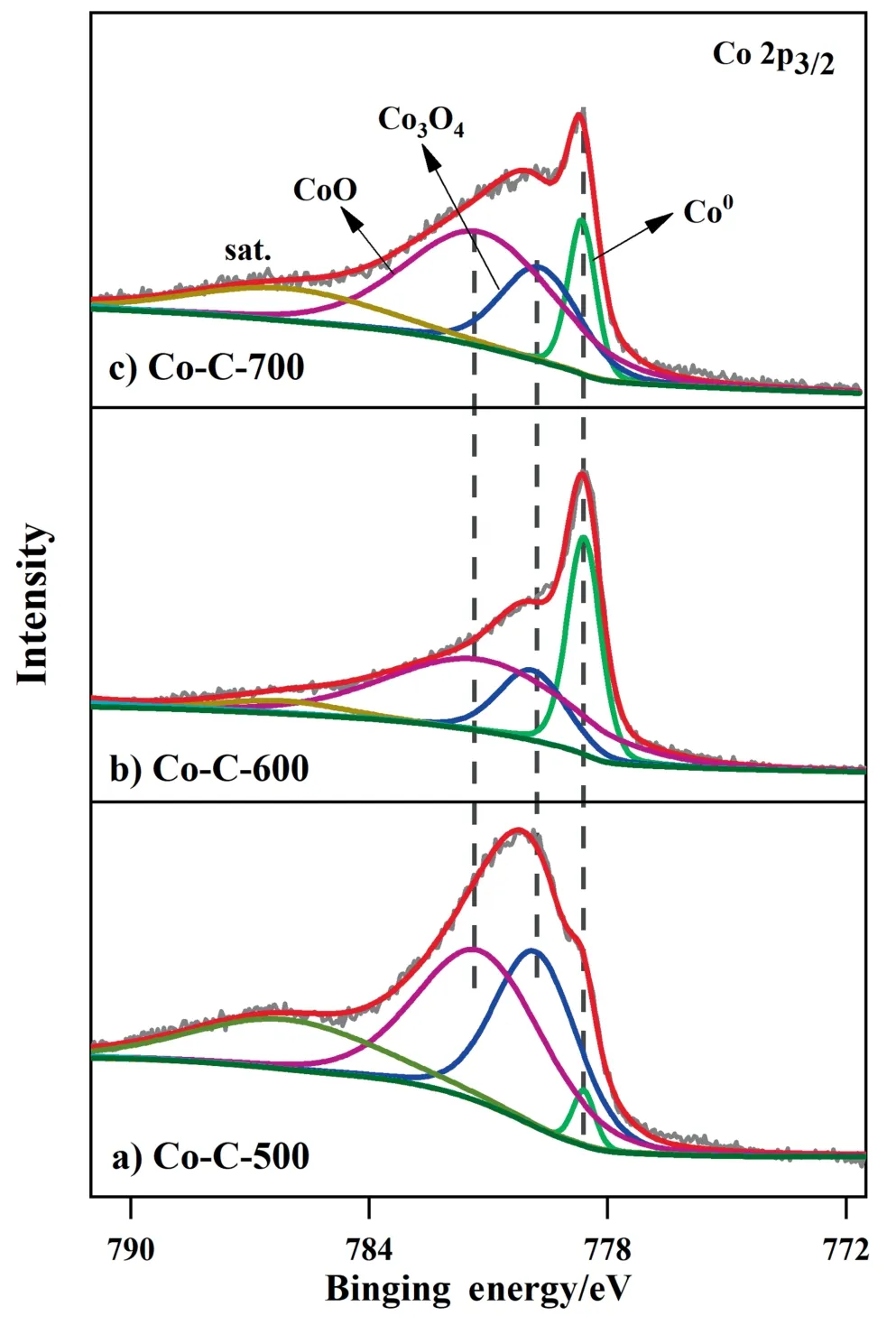
The XPS measurement was employed to investigate the surface chemical states and contents of Co and C specie in Co-C-T.As illustrated in Fig.6,the high-resolution Co 2p3/2XPS spectra showed four peaks,which can be assigned to Co(0) (778.6 eV),Co3O4(779.8 eV),CoO (781.2 eV),and the satellite peak (786.1 eV),respectively [40–43].It can be observed from the data in Table 2 that the contents of Co calculated from the XPS data were lower than those from the ICP-AES results,at the same time,the C contents increased with the carbonization temperature,suggesting that Co species were embedded in the carbon layer.It was also found that the amount of metallic Co was much less than that of CoOxon the surface for the three catalysts(detected by XPS),which is quite different from the XRD results (metallic Co was in dominant amount in the bulk phase).This discrepancy may be explained by the fact that the metallic Co is very active and prone to be oxidized on the surface for air exposure.
3.3.Catalytic performances of Co-C-T in the selective hydrogenation of CAL
3.3.1.Selection of catalysts and optimization of hydrogenation reaction conditions
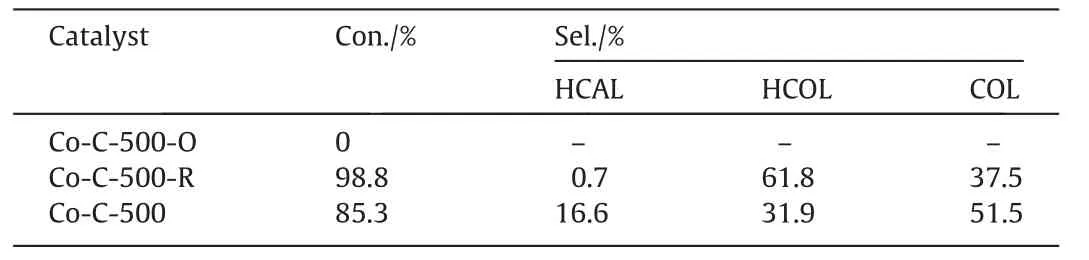
First,the catalytic activity of Co-C-Tfor CAL hydrogenation was preliminarily screened under a set of reaction condition(1.5 mmol CAL,20 mg cat.,10 ml IPA,2.0 MPa H2,120°C,4 h,900 r∙min-1).As can be seen(Fig.7(a)),Co-C-400 displayed the worst catalytic performance and no products were detected;Co-C-500 showed evidently improved catalytic performance,the conversion of CAL reached 70.7%,and the selectivity to COL was 34.8%;however,when the carbonization temperature was further increased,the CAL conversion over Co-C-600 and Co-C-700 decreased to 49.7%and 41.5%,respectively,which may be caused by the agglomeration of Co NPs,the decrease in the specific surface area,and the reduction in the amount of the carbon defect sites as the carbonization temperature increased.Therefore,considering the catalytic activity,Co-C-500 was selected as the optimal catalyst for CAL hydrogenation.
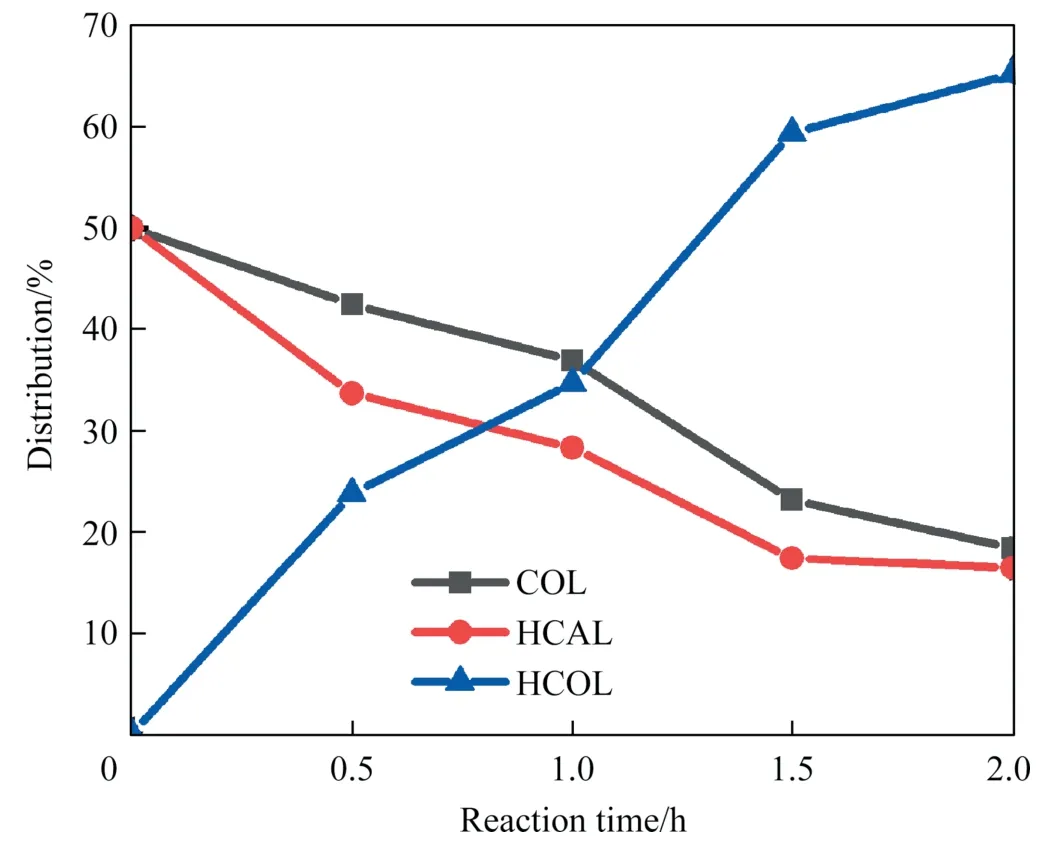
In order to further optimize the reaction conditions of CAL hydrogenation over Co-C-500,a series of experiments were carriedout.Firstly,CAL hydrogenation over Co-C-500 was investigated in several organic solvents with different polarities (polarity order:cyclohexane Table1 The textural properties of Co-C-T Fig.5. SEM images of(a,b)Co-C-500,(c)Co-C-600 and(d)Co-C-700;TEM images with corresponding particle distribution:(f)Co-C-500,(g)Co-C-600,(h)Co-C-700;and(e)HRTEM image and SAED (inset) of Co-C-500. Fig.6. High resolution of Co 2p XPS spectrum of Co-C-T catalysts:(a)Co-C-500,(b)Co-C-600,(c) Co-C-700. Hydrogenation experiments have shown that the catalytic activity of Co-C-500 is closely related to the polarity of the solvent.Meanwhile,it has been reported that the addition of water to the solvent can significantly improve the catalytic activity of the catalyst [44].Therefore,different proportions of water were added to ethanol to test the activity of Co-C-500 in CAL hydrogenation under mild reaction conditions (90 °C,2 MPa H2,4 h).The results are shown in Fig.7(c).When 10%(vol)of water was added,the conversion of CAL evidently increased from 37.3%to 85.3%,showing a significant improvement in the catalytic activity of Co-C-500.When 20%(vol)of water was added,both CAL conversion and COL selectivity decreased.When 10 ml pure H2O was used as solvent,the selectivity to COL was slightly improved under the same reaction conditions,but the conversion of CAL was only 66.0%.This may be due to the poor solubility of CAL in water,which hinder the hydrogenation process [18].Therefore,ethanol–water solution with 10% (vol) of water was determined as the solvent in the consequent experiments. As shown in Fig.7(d),the effect of reaction temperature on CAL hydrogenation was investigated under the reaction conditions of 2 MPa H2,4 h,and the mixture solvent of EtOH and water(10% (vol) of water).In the range of 70–90 °C,the CAL conversion increased significantly with the increase of the reaction temperature,and the selectivity to COL was maintained at 51%–52%.When the temperature was increased to 100 °C,CAL wascompletely converted,but the selectivity to COL decreased significantly to 22%.Comprehensively considering the CAL conversion and the selectivity to the target product COL,90 °C was determined as the optimal reaction temperature.Fig.7(e) and (f) illustrates the effect of reaction time and hydrogen pressure.The conversion of CAL increased as the reaction time prolonged,while the selectivity to COL decreased slowly,at the same time,the selectivity to deep hydrogenation product HCOL increased gradually.When the reaction time was prolonged to 5 h,the selectivity to HCOL increased greatly and became the main hydrogenation product.Therefore,considering the conversion of CAL and the selectivity to the target product COL,4 h was chosen as the moderate reaction time.Similarly,2 MPa H2was selected as the suitable hydrogen pressure.The above investigation suggested the optimal reaction conditions (90 °C,2.0 MPa H2,9 ml EtOH+1 ml H2O,4 h,900 r∙min-1),under which 85.3% of CAL was converted with 51.5% selectivity to COL.Compared with other cobalt-based catalysts reported in the literatures[13,18,30,45–51] (Table 3),the catalyst Co-C-500 showed a higher catalytic activity in CAL hydrogenation and a moderate COL selectivity at a relatively lower reaction temperature. Table2 The contents of Co and C of Co-C-T 3.3.2.Storage stability and reusability of Co-C-500 in the selective hydrogenation of CAL The storage stability and reusability of a heterogeneous catalyst are extremely important indicators for its practical application. The catalyst Co-C-500 was placed in a desiccator for several days on purpose.After that,its catalytic performance was evaluated under a fixed condition.The results are shown in Fig.8(a).Co-C-500 placed for 1 d exhibited nearly identical catalytic performance as the newly prepared one.However,Co-C-500 showed decreased CAL conversion from 85.3% to 77.9% after 3 d,and then to 66.8%after 7 d.That is,a decline by 22%in the catalytic activity was observed after 7 d of placement,indicating a certain storage stability of Co-C-500.To clarify the reason,the catalyst Co-C-500 placed for 1 d and 7 d was characterized by XRD (Fig.8(b)).It can be seen that the peak intensity ascribed to CoOxincreased at the price of the peak intensity attributed to metallic Co after 7 d,which may arise from the slow oxidation of Co(0) by O2in air.The decrease in the metallic Co content in Co-C-500 may be responsible for the decline in the catalytic activity. In addition,when the catalyst Co-C-500 was used in five successive cycles without extra treatment,the CAL conversion and COL selectivity only fluctuated slightly,implying that the catalyst showed excellent reusability (Fig.9(a)).The XRD pattern of Co-C-500-cycled is exhibited in Fig.9(b),metallic Co was the dominant species,accompanied with a small amount of CoOx,which is in agreement with that of the fresh Co-C-500.The TEM image of Co-C-500 after five cycles is displayed in Fig.9(c).A mean particle size of Co around 11.9 nm was observed,almost the same as the fresh catalyst (11.3 nm).That is,no agglomeration and oxidation of the active components in Co-C-500 were observed after 5 cycles,which makes it maintain stable catalytic performance during the cycle testing. Fig.9. (a) Reusability test of Co-C-500 (black line: conversion,histogram: selectivity);reaction conditions: CAL 1.5 mmol,Co-C-500 20 mg,EtOH 9 ml,H2O 1 ml,hydrogen pressure 2.0 MPa,90 °C,4 h,900 r∙min-1.(b) XRD of fresh and cycled Co-C-500;(c) TEM image with corresponding particle distribution of cycled Co-C-500. Table4 Control experiments to confirm the hydrogen source Fig.10. XRD patterns of Co-C-500 (a) before and (b) after oxidation. 3.3.3.Study on active components It has been reported that proton alcohols can often be used as hydrogen donors in Co-based catalyzed hydrogenation reactions,and hydrogen transfer reactions often take place in inert atmosphere [52,53].For example,Wanget al.[54] established an efficient method for transfer hydrogenation of CAL to COL usingethanol as solvent and hydrogen donor.Therefore,a control experiment was carried out to clarify the source of hydrogen supply for the reaction.When 2 MPa N2rather than 2 MPa H2was introduced into the reaction system,no CAL conversion was detected under the optimal reaction condition(Table 4),indicating that H2is the only source of hydrogen in the catalytic system. Table5 Catalytic performance in selective hydrogenation of CAL over Co-C-500 before and after oxidation/reduction Since two states of Co species were detected in the catalyst Co-C-500: oxidized CoOxand metallic Co(0),experiments are designed to determine the active component for CAL hydrogenation.First,the freshly prepared Co-C-500 was subjected to slow oxidation at 200 °C in a muffle furnace for 1 h.The XRD results(Fig.10) of the oxidized Co-C-500 (denoted as Co-C-500-O)confirmed that most of the metallic Co species in the catalyst were oxidized to CoOx.As a control,the precursor Co-BTC was carbonized in 5% H2/Ar atmosphere at 500 °C for 2 h to obtain Co-C-500-R.The catalytic activities of Co-C-500-O and Co-C-500-R were evaluated in CAL hydrogenation,and the results are listed in Table 5.As can be seen,almost no CAL conversion was detected on Co-C-500-O,while 98.8% of CAL conversion was achieved on Co-C-500-R,indicating that Co(0) acts as the active component for CAL hydrogenation. In order to compare the independent hydrogenation rates of C=C double bond and C=O double bond on Co-C-500,a mixture containing equimolar ratio of HCAL (only containing C=O double bond) and COL (only containing C=C double bond) was used as the reactant.As shown in Fig.11,the content of HCOL,the full hydrogenation product,increased with the reaction time.And it can be found that the hydrogenation rate of HCAL was slightly higher than that of COL,indicating that the hydrogenation rate of C=O bond was a little bit higher than that of C=C bond.That is,the C=O bond is somewhat preferentially adsorbed and activated on Co-C-500,thus COL was the main product of CAL hydrogenation. Fig.11. Kinetic investigation of the hydrogenation the mixture containing equimolar HCAL and COL on Co-C-500.Reaction conditions: 1.5 mmol HCAL and 1.5 mmol COL,Co-C-500 20 mg,EtOH 9 ml,H2O 1 ml,hydrogen pressure 2.0 MPa,90 °C,900 r∙min-1. To sum up,the catalysts Co-C-Twere successfully synthesized through one-step carbonization of Co-BTC precursor.It was found that the carbonization temperature affected the number of defect sites of the carbon support,the size of cobalt nanoparticles,and the pore structure of the catalysts.The optimal catalyst Co-C-500 exhibited 85.3% conversion of CAL and 51.5% selectivity to COL for the hydrogenation of cinnamaldehyde,under the conditions of 90 °C,4 h,2 MPa H2,using 9 ml ethanol and 1 ml water as the solvent.The high activity of Co-C-500 at relatively low temperature can be ascribed to the uniformly dispersed metallic cobalt nanoparticles and the large specific surface area of the catalyst.Moreover,Co-C-500 showed considerable placement stability and excellent reusability due to the metal cobalt nanoparticles embedded in carbon carrier.The high catalytic activity of Co-C-500 in CAL hydrogenation shown in this work demonstrates that Co based catalyst could be an effective candidate in the selective hydrogenation of α,β-unsaturated aldehydes. Data Availability Data will be made available on request. Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Acknowledgements The authors are grateful for financial support from the National Natural Science Foundation of China (22272016).




4.Conclusions
 Chinese Journal of Chemical Engineering2023年10期
Chinese Journal of Chemical Engineering2023年10期
- Chinese Journal of Chemical Engineering的其它文章
- High catalytic performance of CuCe/Ti for CO oxidation and the role of TiO2
- Experimental and numerical studies of Ca(OH)2/CaO dehydration process in a fixed-bed reactor for thermochemical energy storage
- Volumetric and ultrasonic properties of thiamine hydrochloride drug in aqueous solutions of choline-based deep eutectic solvents at different temperatures
- Synthesis of zeolite A and zeolite X from electrolytic manganese residue,its characterization and performance for the removal of Cd2+ from wastewater
- A pseudo transient nonequilibrium method for rigorous simulation of multicomponent separation columns
- Recent development of catalytic strategies for sustainable ammonia production