
自噬联合其信号通路在骨骼肌衰老中的研究进展*
2023-12-24 05:15王京峰文登台王士杰高颖晖
宝鸡文理学院学报(自然科学版) 2023年3期
王京峰,文登台,王士杰,高颖晖
(鲁东大学 体育学院,山东 烟台 264025)
自噬在进化过程中是高度保守的,从酵母和果蝇到脊椎动物和人类都可以找到参与自噬的同源基因。其命名法最初被称为APG,AUT和CVT,现在已经统一为酵母自噬相关基因Atg。哺乳动物自噬基因的命名法与酵母相似,但也有个别差异。如酵母的Atg1,6和8在哺乳动物中分别被称为ULK1,Beclin1和LC3。细胞自噬是一个细胞内的回收系统,需要内源性底物和细胞器的转换[1]。自噬泡的形成大小受到细胞内外氨基酸浓度和ATP水平的严格调节,这些信号通路包括Atg家族和mTOR激酶[2]。已经确定的3种类型的自噬(微自噬、伴侣介导的自噬和巨自噬)见图1。
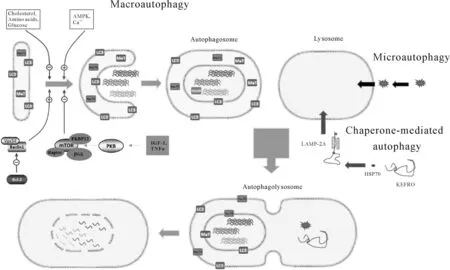
图1 自噬过程及其分类Fig. 1 The autophagic process and its classification
通过形成具有双层膜结构的自噬体来包裹细胞内物质,自噬体最终与溶酶体融合。一般来说,自噬是指宏观自噬。AMPK信号通路的激活和Ca++的参与促进了巨量自噬,而胆固醇、氨基酸、葡萄糖和其他此类物质会减弱或抑制这一过程。蛋白质如Beclin1和Vps34可以促进巨噬细胞的过程,而Bcl-2可以减弱或抑制Beclin1和Vps34的作用。IGF-1和TNF是PKB的上游调节器,它们的正常作用可以促进其生理表达,而生理表达又可以调节mTOR,FKBP12,GL和Raptor来减弱或抑制巨噬细胞的过程。 微自噬通过溶酶体或囊泡表面的变形直接吞噬特定的细胞器。分子伴侣介导的自噬在HSP70伴侣的帮助下,具有KEFRQ类的蛋白质通过LAMP-2A转运器被运送到溶酶体。
和细胞凋亡与衰老一样,细胞自噬是一种非常重要的生物现象,受损的大分子和细胞器通过细胞自噬来降解。它是一种重要的细胞应激反应,在缺氧和营养缺乏时提供生物能量的中间体[3]。自噬在延长寿命的不同模式中是必需的[4]。正常水平的自噬对持续的细胞更新至关重要。基础自噬在维持细胞平衡方面发挥着重要作用,而过度自噬则可能导致自噬性细胞死亡。自噬的改变在包括癌症、神经退行性疾病、衰老、代谢性疾病、炎症和心血管疾病等疾病中起着重要作用[5]。
1 自噬与骨骼肌的关系
自噬在骨骼肌中持续活跃,在组织稳态中的作用很复杂。许多研究已经证明了自噬在肌肉生长、萎缩、肥大、再生和运动过程中的作用。抑制自噬会导致泛素化蛋白在各种细胞类型中的积累[6],包括肌肉细胞以及线粒体、内质网以及高尔基体的异常[7]。自噬作用与肌肉再生有关,决定了肌肉干细胞从静止到衰老的命运。研究表明,卫星细胞需要结构性自噬来维持其干细胞的适应性。衰老的卫星细胞或有基因缺陷的细胞(如Atg7基因敲除小鼠)的自噬失败导致衰老、氧化应激和线粒体功能障碍,以及细胞器和蛋白质的积累[8]。自噬的重新激活可以恢复其活性。在Atg7基因中,第14号外显子的核苷酸编码了一个半胱氨酸残基,这是激活底物所必需的。成年小鼠肝脏特异性靶向删除Atg7的14号外显子,破坏了自噬泡的形成,抑制了禁食条件下蛋白质的整体降解。此外,肌肉特异性Atg7敲除小鼠表现出异常的线粒体、无序的肌浆、网状扩张和异常的同心膜结构。它们表现出肌肉表型,如肌肉损失和分解代谢条件下的变性。这些数据表明,在生理条件下,基础水平的自噬在维持肌肉质量和肌肉纤维完整性方面发挥了有益的作用。与LAMA2基因突变相关的过度自噬导致了大量的肌肉萎缩[9]。小鼠层粘连蛋白α2链缺失导致肌肉中自噬相关基因的表达增加,并导致肌肉纤维化、萎缩和凋亡表型,表明过度自噬的病理作用[10]。可见,静态自噬的破坏导致了各种肌肉疾病的发病机制,骨骼肌中的自噬水平需要得到精细的调节和适当的调控,以维持骨骼肌在生理和应激条件下的稳态。骨骼肌疾病与自噬有关(表1)。

表1 与自噬有关的骨骼肌疾病、表现特征以及成因Tab. 1 Skeletal muscle diseases associated with autophagy, presenting features, and causes
2 控制骨骼肌衰老的自噬的信号通路
在骨骼肌中,自噬受到多种信号输入的严格调节,并与其他信号通路相互作用(图2)。

图2 控制骨骼肌衰老的自噬分子途径Fig. 2 Molecular pathways of autophagy that control skeletal muscle aging
2.1 AMPK
AMPK是代谢抑制的主要传感器,由细胞ATP水平的下降(腺苷/ATP比率的增加)激活[11]。AMPK作为一个能量传感器,监测细胞内ATP水平的变化,这在高能量消耗的肌肉中特别重要。骨骼肌的生长取决于蛋白质分解代谢和合成代谢之间的平衡。当肌肉蛋白分解率高于肌肉蛋白合成率时,就会发生萎缩。AMPK被升高的AMP/ATP比率激活,上调ATP的分解代谢途径,同时抑制ATP消耗的生物合成过程[12]。在成熟的骨骼肌中,LKB1(肝脏激酶B1)通常被认为是主要的AMPK激酶,因为肌肉特异性敲除LKB1基本上可以消除总的AMPK活性。然而,LKB1似乎在激活AMPKα2方面起着更重要的作用,因为敲除LKB1并不严重影响骨骼肌中AMPKα1的活性[13]。CamKK(钙/钙蛋白依赖性蛋白激酶)和TAK1(转化生长因子β激活的激酶-1)也可能在某些条件下激活骨骼肌的AMPK中发挥重要作用[14]。
节诱导自噬相关基因LC3B-II,Gabarapl1和BECLIN1的表达[15]。然而,值得注意的是,尽管AMPK在已知激活转录因子的位点上磷酸化FoxO3a,从而诱导广泛的蛋白质降解,但这并不一定影响其在细胞核中的定位[16]。AICAR(AMPK激活药物)也增加FoxO3与MuRF-1,Atrogin-1启动子的结合[17]。此外,AMPK的激活增加了NAD+(烟酰胺腺嘌呤二核苷酸)的浓度,从而激活了Sirt1(sirtuin 1)的去乙酰化酶。Sirt1介导的FoxO蛋白的去乙酰化增加了其转录活性[18]。此外,在营养饥饿条件下,哺乳动物AMPK直接磷酸化ULK1的Ser 317和Ser 777,促进自噬的启动,并随后提供能量和营养物质[19]。在AMPK激活后,由AMPK,mTORC1,ULK1,FIP200和Atg13组成的复合物会释放ULK1,从而导致自噬的激活[20]。这些结果表明,在正常的营养条件下,需要基础水平的自噬来降解错误折叠的蛋白质和受损的细胞器,以维持平衡。相反,在饥饿和运动等应激反应下,AMPK的激活可以上调自噬,以降解蛋白质作为营养和能量的替代来源[15]。在能量丰富的条件下,mTORC1通过Ser757位点的磷酸化抑制ULK1。这与mTOR靶向其他自噬成分一起,导致了mTOR对自噬的抑制[21]。近几十年来,AMPK作为控制能量平衡的细胞过程的信号通路的作用已得到充分肯定。尽管它肯定不是调节骨骼肌发育、大小和/或生长的唯一角色,但它,特别是AMPKα1亚单位,已经成为限制肌肉大小和肥大能力的关键因素。另一方面,AMPKα2可能在通过自噬和蛋白质降解促进肌肉萎缩方面发挥比AMPKα1更重要的作用[22]。
2.2 mTORC1
mTORC1由Raptor,mLST8,PRAS40和DEPTOR组成,对雷帕霉素敏感[23]。mTORC1的激活是通过与GTP结合的Rheb相互作用在溶酶体膜上进行的[24]。Raptor(相对于mTORC2)的基因表达量较低,而Raptor是mTORC1的定义成分,但PRAS40的基因表达量较高,这意味着mTORC1途径与人类寿命之间存在负相关关系[25]。鉴于mTORC1是一种关键的营养感应激酶,在生长条件下对氨基酸和葡萄糖有抑制自噬作用[26],抑制mTORC1可能有助于限制热量的抗衰老作用。
一些研究表明,mTORC1信号通路对肌肉功能至关重要。TSC2(肌肉特异性结节性硬化症复合体2)联合TSC1,TBC1D7(TBC1结构域家族成员7)作为GTP酶激活蛋白,将GTP转化为GDP,从而大大降低Rheb促进mTOR活性的能力[27]。 缺乏TSC的小鼠在喂养、基础和饥饿条件下显示出mTORC1的持续激活和LC3Ⅰ和LC3Ⅱ的水平不变,这表明饥饿依赖的自噬受阻。这种受损的自噬作用导致了严重的晚期肌病。AMPK通过多个步骤对mTORC1途径进行负向调节。它磷酸化并激活mTOR的上游抑制剂TSC1/2,同时也磷酸化Raptor,两者都能抑制mTORC1。此外,AMPK直接磷酸化并激活ULK1和Beclin1以诱导自噬[19]。雷帕霉素治疗可恢复自噬并减弱小鼠的肌病表型。尽管FoxO3被激活,但构成性自噬和饥饿诱导的自噬被mTORC1介导的对ULK1的抑制所阻断。mTORC1将ULK1的几个位点磷酸化,如Ser 757,从而阻断ULK1和AMPK之间的相互作用[28]。矛盾的是,尽管自噬基因的FoxO依赖性转录减少,但通过缺失消除mTORC1的活性也能诱导自噬。这些数据表明,mTORC1是诱导骨骼肌自噬的另一个上游调节器[29]。
2.3 FoxO
哺乳动物的FoxO家族由FoxO1,FoxO3,FoxO4和FoxO6蛋白组成,它们在体内广泛表达。它们介导胰岛素或胰岛素样生长因子对细胞代谢、生长、分化、氧化应激、衰老、自噬和衰老的关键功能的抑制作用。FoxO基因的突变或FoxO蛋白的异常表达与人类和动物的代谢性疾病、癌症或寿命的改变有关[30]。FoxO是重要的转录因子之一,对自噬相关的转录因子以及溶酶体基因的表达有积极的调节作用[31]。众所周知,泛素介导的蛋白酶体系统(UPS)和自噬/溶酶体系统是降解受损或错误折叠的蛋白质的2个主要机制。研究发现,自噬溶酶体和泛素蛋白酶体系统都受到FoxO蛋白的控制,导致骨骼肌蛋白质流失。FoxO家族的成员调节萎缩相关的泛素连接酶atrogin1/MAFbx,MuRF1,TRIM63,MUSA1,SMART,UBC,USP14和Ube4b,以及其他编码蛋白体亚单位的基因,这些基因共同参与了肌肉萎缩[32]。
FoxO3在骨骼肌中作为自噬基因ATG4,ATG8B,ATG12,LC3,BECLIN1,BNIP3,VPS34,ULK1和ULK2转录的激活剂发挥作用[33]。重要的是,FoxO3通过自噬在肌肉萎缩中发挥了必要的作用。抑制Bnip3很大程度上阻断了FoxO3诱导的自噬作用[33]。一项使用肌肉特异性FoxO1,3,4/小鼠的研究发现,在禁食条件下,63个萎缩相关基因中有29个被FoxO控制。FoxO1,3,4/小鼠中自噬相关基因如LC3,Gabarapl和Bnip3的诱导被抑制。在其他与自噬相关的途径中起作用的基因,如未折叠蛋白反应,在FoxO1,3,4/小鼠中也在应激条件下受到负面影响[32]。这些结果同样也支持FoxO转录因子在肌肉萎缩中的调节作用。
2.4 核因子-κB(NF-κB)
已有数百个基因被确定为受NF-κB转录控制[34]。最近的研究表明,在不同的生理和病理条件下,NF-κB的激活与骨骼肌质量的损失有关。发现骨骼肌特异性删除IKKβ(NF-κB信号传导的上游激活剂)可抑制MuRF1 E3泛素连接酶的表达。NF-κB抑制蛋白IκBα的显性负突变体过量表达也可抑制肌肉中的蛋白降解[35]。MyD88的靶向消融抑制了去神经化过程中骨骼肌中的泛素蛋白酶体系统、自噬和FoxO转录因子的成分。而对MyD88的特异性抑制能减少典型的核因子κb(NF-κB)途径的激活和去势肌肉中炎症因子受体的表达。相反,对MyD88的抑制刺激了非典型的NF-κB信号通路在去神经化的骨骼肌中的激活[36]。一些报道的结果支持NF-κB在肌形成前的作用。研究发现,IGF-Ⅱ(胰岛素生长因子Ⅱ)刺激NF-κB的激活,这反过来又诱导了成肌信号传导途径[37]。这些对肌肉的抗或促成肌作用可能是由典型和非典型的NF-κB信号途径之间的转换决定的[38]。这些结果表明,NF-κB的激活在肌肉萎缩中起着重要作用。
NF-κB蛋白是一个转录因子家族,通过转录诱导编码凋亡和自噬细胞死亡蛋白的基因,在调节细胞死亡方面发挥重要作用。许多研究描述了NF-κB和细胞自噬之间的关系。NF-κB信号已被证明以环境依赖的方式参与自噬[39]。已有研究表明,lps诱导的NF-κB激活及其下游介质(如COX-2,IL-1β,TNF-α和NLRP3)在骨骼肌萎缩的过程中发挥了重要作用[40]。促炎症细胞因子TWEAK通过Traf6介导的NF-κB激活几个自噬基因的表达,包括BECLIN1,LC3B和Atg5,促进骨骼肌萎缩[41]。在相关的信号通路中,NF-κB是维持骨骼肌平衡的一个关键因素。NF-κB以二聚体形式存在,参与各种疾病的发展和进展,如炎症、细胞凋亡和增殖[42]。事实上,NF-κB的激活足以诱导自噬途径相关或关联基因的表达,如BECLIN1和BAG3-HspB8复合物[43]。在饥饿条件下,IKKα和IKKβ也以NF-κb无关的方式刺激Atg5,BECLIN1和LC3的表达[44]。另一方面,NF-κB信号通路在某些条件下对细胞自噬也有抑制作用,这可能是通过间接机制介导的。NF-κB信号通路可能激活mTOR激酶,促进Bflfl-1/A1(Bcl-2家族成员和BECLIN1结合伙伴)等自噬抑制剂的表达,并阻止JNK1,BNIP3,p53和ROS表达等自噬诱导因子的表达[45]。这些研究表明,NF-κB是骨骼肌自噬的一个重要调节器。虽然NF-κB和自噬之间的关系已被深入研究,但这些研究大多集中在癌症方面。NF-κB在肌肉自噬中的作用尚不清楚,需要进一步的研究。
2.5 糖皮质激素受体(GR)
糖皮质激素受体(GR)信号传导已被证明与骨骼肌中的蛋白质合成和蛋白质水解有关。GR信号传导在肌肉细胞蛋白质分解中起着关键作用[46]。肌肉蛋白水解是一个活跃的过程,由特定的信号通路和转录程序严格控制,涉及泛素蛋白体和自噬机制。GR转录控制着这2种机制中各成分的表达水平,并调节着肌肉肝脏脂肪信号轴[47]。在禁食期间,地塞米松被用来激活糖皮质激素受体,地塞米松增加了聚泛素mRNA的表达并激活了骨骼肌中的蛋白水解途径[48]。肾上腺切除的大鼠肌肉蛋白分解的减少可以通过给予糖皮质激素来抵制[49]。这些结果表明,GR信号是骨骼肌蛋白分解所必需的。已发现糖皮质激素在肌肉萎缩中刺激FoxO1和3的mRNA并激活UPS相关蛋白的表达,如atrogin1,Murf1和Fbxo30[50]。对照组和糖皮质激素受体敲除组(GRKO)小鼠在缺氧和减食条件下都能诱导自噬基因Map1lc3b和Bnip3的表达,但GRKO小鼠没有反应,且FoxO1表达受损。同样在小鼠模型中,有研究发现在常氧条件下,由于食物摄入减少,与肌肉萎缩和蛋白质水解有关的基因表达增加需要GR信号。在缺氧条件下,肌肉萎缩和泛素蛋白体系统相关的E3配体Murf1和Atrogin-1的基因表达升高大多与GR信号传导无关。此外,在缺氧诱导的肌肉萎缩中,对mTORC1活性的抑制受损是GR依赖性的[48]。此外,mTOR抑制因子REDD1和KLF15已被确定为糖皮质激素的直接靶基因。Redd1的表达可诱导自噬并抑制蛋白质的合成[51]。kLF15还激活了诱发骨骼肌萎缩的atrogin-1和MuRF1基因的表达。这些数据表明,糖皮质激素受体信号通路可能通过控制基因表达在肌肉萎缩的情况下促进自噬[52]。众所周知,肌肉萎缩会降低身体功能和整体健康。糖皮质激素分泌增加或使用处方糖皮质激素可通过激活糖皮质激素受体而明显诱发肌肉萎缩,糖皮质激素受体转录改变蛋白质平衡的基因,有利于蛋白质降解[53]。因此,利用上述的一套机制来有效地调节糖皮质激素将是一项有趣的研究。
2.6 转化生长因子(TGF-β)
TGF-β信号传导是各种生理过程中最重要的信号传导途径之一。在一项关于益生菌对体外纤维化的影响的研究中,发现TGF-β不仅能促进胶原蛋白的沉积,还能显著上调LX-2细胞中Col1A1,α-SMA(α-平滑肌肌动蛋白)、MMP-2(基质金属蛋白酶-2)、IL-6,CXCL-8,CCL2和IL-1β的mRNA水平[53]。自噬可能与TGF-β信号传导呈负相关。最近的一项研究发现,前列腺素降解酶15-PGDH在肌肉中随年龄增长而升高,这可能导致与年龄有关的肌肉萎缩。通过小分子或基因缺失抑制15-PGDH对肌肉恢复活力有好处,这是由一系列事件介导的,如PGE2的增加、线粒体功能的恢复、促生长基因表达和TGF-β信号的减少以及自噬的增加[54]。然而,一项相关研究的结果也预测,自噬可能与TGF-β信号传导呈正相关。 c-Ski是TGF-β信号传导的抑制剂,参与各种生理和病理过程。ox-LDL(氧化的低密度脂蛋白)和PDGF(血小板衍生的生长因子)直接诱导c-Ski蛋白的降解,并与它的磷酸化减少密切相关。S383(丝氨酸383)被认为是稳定c-Ski蛋白表达和核定位的关键磷酸化位点,并与自噬相关基因的转录抑制有关。S383磷酸化水平的降低促进了c-Ski降解,从而削弱了其对自噬基因诱导的抑制作用[55]。因此,通过目前的研究结果,自噬和TGF-β信号之间的关系仍然是模糊的,但有一点可以肯定的是,在骨骼肌中,TGF-β介导的作用与自噬环境有关。
GDF-8(生长/分化因子-8,肌肉生长抑制剂)是TGF-β家族的成员,是一种从早期胚胎发育到成年的骨骼肌特异性蛋白。GDF-8基因的缺失会导致骨骼肌质量增加3倍,这是由增生或肥大引起的[56]。在人类和哺乳动物中已经发现了GDF-8基因的多种突变,这导致了肌肉质量的增加[57]。在小鼠和人类中,肌肉生长抑制物的水平随着基因的异变导致的肌肉萎缩而增加,以及癌症恶病质、慢性心力衰竭、慢性阻塞性肺病、艾滋病和糖尿病患者[58]。通过肌肉生长抑制剂和eIF2α磷酸化的信号通路导致蛋白质合成和mTORC1活性下降,同时伴随线粒体功能障碍和自噬增加,导致肝病骨骼肌高氨血症期间蛋白酶稳定性紊乱[59]。高氨血症导致的肌肉生长抑制因子的上调是造成肌肉疏松症中蛋白酶稳定性受损的原因。在骨骼肌中,氨通过Rh-B糖蛋白样氨转运体进入肌细胞。高氨血症通过NF-κB依赖的受体独立途径导致肌肽的表达[60]。GDF8与其受体的结合导致了转录因子SMAD2/3的磷酸化和SMAD4的招募。SMAD2/3的激活抵消了JunB对FoxO3的抑制作用,导致了萎缩[61]。SMAD3的表达也增加了atroin-1,MuRF1和PGC1α的启动子活性,激活了PTEN30-UTR和FoxO反应元件报告因子,并抑制了miR-29启动子的活性和骨骼肌中的mTOR,导致了蛋白分解和骨骼肌纤维萎缩[62]。这些结果表明,GDF8在肌肉萎缩中起着关键作用。
2.7 泛素蛋白酶体系统(UPS)
蛋白质水解是骨骼肌修复和再生所必需的。溶酶体介导的自噬和泛素蛋白酶体系统是细胞内主要的肌肉蛋白降解系统。这2个系统通过不同的机制识别泛素化,是降解的信号和系统之间的动态互动。越来越多的证据表明,自噬和泛素蛋白体系统在各种病理条件下的骨骼肌萎缩进展中是相互依赖的[63]。特别是UBE2D3,它是泛素结合酶E2家族之一,已被证明能促进p62/SQSTM1泛素化,导致心肌I/R损伤诱发的自噬通量损伤增加。此外,UBE2D3也可能通过负调控mTOR参与调节自噬。然而,更令人惊讶的是,这种机制独立于已知的mTOR-beclin1途径[64]。过去20 a的研究揭示了骨骼肌中这2种途径之间的联系。转录因子FoxO3已被证明能刺激许多自噬相关基因的表达,而E3泛素连接酶atrogin-1/MAFbx则能刺激自噬相关基因的表达,以及肌肉中溶酶体蛋白的水解。抑制自噬会导致肌肉萎缩以及肌肉质量的损失,与破坏atroin-1和MuRF1(2个与萎缩相关的泛素连接酶)的功能以及删除参与不同分解代谢途径的基因所造成的表型相似[65]。因此,自噬溶酶体途径和泛素蛋白酶体途径在FoxO调节的下游平行发挥作用,并可能相互补充。
3 结果与结论
骨骼肌是人体中最丰富的组织类型,在分解代谢和合成代谢过程中保持着良好的代谢平衡。自噬是一个重要的分解代谢过程,负责通过自噬体溶酶体系统降解异常的蛋白质和细胞器,进化保守的Atg蛋白参与其中。自噬的激活可以介导许多延缓衰老的干预措施,其过程需要多种信号通路的参与,在维持细胞内平衡方面发挥着核心作用。异常的自噬水平可能会导致细胞损伤,引起病理连锁反应,并且出现肌肉无力和萎缩等症状。另外,自噬基因的突变和自噬途径的失调已被确定为各种肌肉疾病的主要原因之一。尽管过去20 a来,人们在了解自噬在骨骼肌衰老中的作用方面取得了进展,但关于骨骼肌衰老中自噬活动异常的分子机制,仍有许多东西需要阐明。在研究其他细胞和组织类型时采用的传统实验技术可以进一步用于研究自噬对骨骼肌衰老的作用,特别是研究这种作用是否具有细胞类型和组织类型的特异性。此外多组学技术取得了惊人的进步,并被广泛地应用于复杂的人类疾病研究。基因水平的测序以及蛋白质组学的研究方法为探究骨骼肌衰老分子水平的机制开阔了视野。从我们回顾的文献中可以看出,骨骼肌衰老显然与细胞代谢有关。自噬的变化及其与其他信号通路的相互作用不仅反映在基因表达上,也反映在蛋白质水平翻译后的修饰上。多组学技术的应用将使人们更广泛地了解自噬对骨骼肌衰老的影响,更深入地了解自噬对疾病发病机制的影响。
猜你喜欢
肝博士(2022年3期)2022-06-30
海外星云(2021年9期)2021-10-14
中国运动医学杂志(2016年3期)2016-07-10
中国运动医学杂志(2016年3期)2016-07-10
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
医学研究杂志(2015年5期)2015-06-10
中国医学科学院学报(2015年5期)2015-03-01
现代检验医学杂志(2015年2期)2015-02-06
四川生理科学杂志(2014年3期)2014-02-28
实用老年医学(2013年7期)2013-03-11
