
土壤三普指定方法测砷汞与行业标准比对探讨
2023-12-14 06:03:54杨超杰刘文水
广州化工 2023年14期
杨超杰,刘文水
(1 广东轻工职业技术学院,广东 佛山 528200;2 广东源创检测技术有限公司,广东 广州 511300)
土壤是人们赖以生存、不可或缺的资源,此次土壤普查旨在对我国土壤类型及分布规律、土壤资源现状及变化趋势等进行全面摸底和调查,真实准确掌握土壤质量、性状及利用状况等基础资料。2022年开始,我国已制定好方案,并分发至各个省份,开始收集参与本次普查的企业名单并进行筛选,每个省份选出条件符合的前三十名作为可参与企业,并且同时也在制定采样、分析、质控等要求,确保本次普查能够顺利展开。本次将对砷和汞两种金属元素的测定方法与行业标准进行研究和探讨,以便找出更好、更快、更准确的方法为我国之后的使用方法多一份参考。
1 实 验
1.1 主要仪器和设备
原子荧光光度计、微波消解仪、可控温水浴锅、可控温电热板。
1.2 主要试剂和材料
砷标准溶液(1 000 mg/L)、汞标准溶液(1 000 mg/L)、盐酸(优级纯)、硝酸(优级纯)、氢氧化钾、硼氢化钾、重铬酸钾、硫脲、抗坏血酸。
1.3 前处理过程
1.3.1 标准
按照国家标准《土壤质量 总汞、总砷、总铅的测定原子荧光法》执行。
(1)砷
取经过0.149 mm过滤筛筛分的土壤0.5 g于50 mL比色管,加入10 mL王水(3份硝酸:1份盐酸),于100 ℃水浴锅中加热2 h,取出冷却。再从消解液中取10 mL于25 mL 比色管,加入盐酸3 mL,硫脲溶液5 mL,抗坏血酸溶液5 mL。定容至25 mL,放置2 h以上后进行测定[1]。
(2)汞
取经过0.149 mm 过滤筛筛分的土壤0.2 g 于50 mL比色管,加入10 mL 王水(3份硝酸:1份盐酸),于100 ℃ 水浴锅中加热2 h,取出冷却,再加入10 mL的保存液,以稀释液定容到50 mL,放置2 h 以上后测定[2]。
1.3.2 行业标准《土壤和沉积物 砷、汞、硒、锑、铋的测定 微波消解/原子荧光法》
称取过0.149 mm 过滤筛筛分的样品0.2 g 置于溶样杯中,用少量超纯水润湿。加入盐酸6 mL 、硝酸2 mL。再将溶样杯置于消解罐中密封。将消解罐装入消解罐支架后放入微波消解仪的炉腔中,按照表1优化好的升温程序进行微波消解,程序结束后冷却[3]。过滤至50 mL 比色管,用超纯水定容。

表1 微波消解仪升温程序优化
测试汞时,在50 mL比色管中取10 mL消解液。加入2.5 mL 盐酸,再定容至50 mL 后待测。测试砷时,在50 mL比色管中取10 mL消解液。加入5 mL 盐酸、10 mL 硫脲-抗坏血酸混合液,定容后待测[4]。
1.4 仪器条件优化
通过反复对原子荧光光度计的调试,设置出该在测量土壤砷和汞时最佳条件如表2所示。
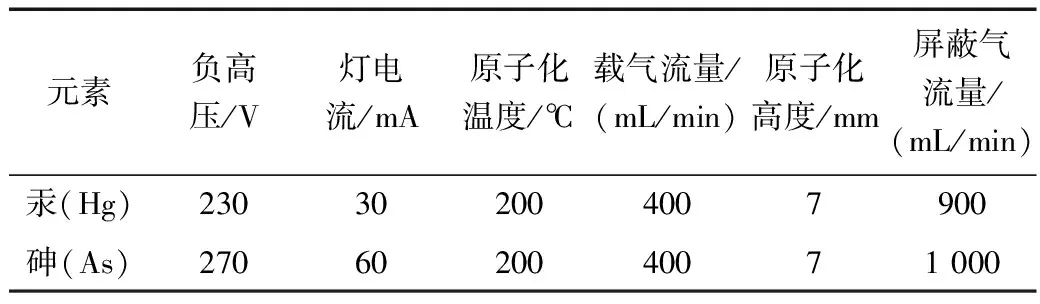
表2 仪器测试条件优化
2 结 果
2.1 检出限
在设置好仪器条件后,根据环境监测分析方法标准制订技术导则(HJ 168-2020)规定,使用全程序空白进行12次测定,并计算其标准偏差,再用3倍标准偏差进行计算检出限[5],结果如表3所示。由表3得出汞的检出限为0.001~0.002 mg/m3,砷的检出限为0.005~0.008 mg/m3,两个标准均适用于土壤砷和汞的检测,但是在前处理时间上,国家标准《土壤质量 总汞、总砷、总铅的测定原子荧光法》(GB/T 22105-2008)所消耗的时间远远大于行业标准《土壤和沉积物 砷、汞、硒、锑、铋的测定 微波消解/原子荧光法》(HJ 680-2013),且在稳定性上,行业标准略优于国家标准。
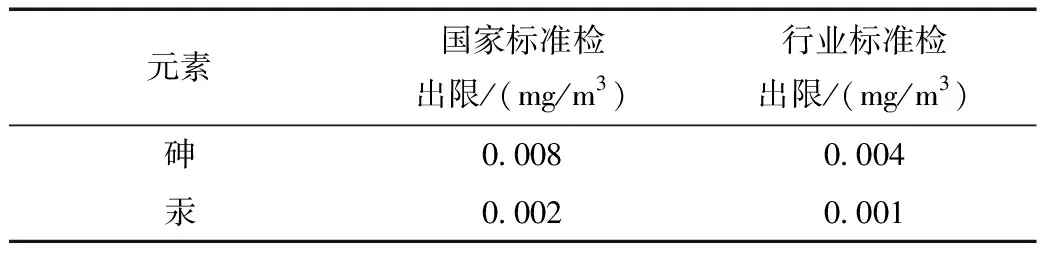
表3 国家标准和行业标准砷汞检出限结果
2.2 精密度
2.2.1 国家标准物质精密度
采用国家土壤标准物质进行测量,国家标准与行业标准分别取7次进行测试,按照对应的前处理步骤进行消解后上机测试,其精密度分析结果如表4所示。由表4可得知其测试结果均在置信区间范围内,但是行业标准《土壤和沉积物 砷、汞、硒、锑、铋的测定 微波消解/原子荧光法》(HJ 680-2013)的精密度明显要高于国家标准《土壤质量 总汞、总砷、总铅的测定原子荧光法》(GB/T 22105-2008)。两者的相对标准偏差分布在0.7%~5.4%以内。
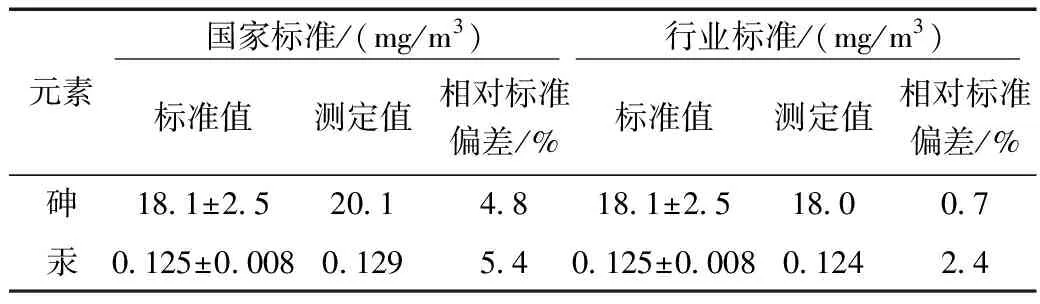
表4 国家标准物质精密度结果
2.2.2 实际样品精密度
于某工业园挖取其表层土,使用国家标准《土壤质量 总汞、总砷、总铅的测定原子荧光法》(GB/T 22105-2008)和行业标准《土壤和沉积物 砷、汞、硒、锑、铋的测定 微波消解/原子荧光法》(HJ 680-2013)进行更深层次的一次比较,分别取7次样品,按照前处理过程对实际样品进行消解后测定,得出其结果如表5所示,其相对标准偏差分布在1.2%~7.5%间,实验过程中可看出国家标准的前处理步骤不仅消耗时间长,且消解过程中容易喷盖导致受污染,或者消解水温稳定影响结果测定。而行业标准前处理使用微波消解法能避免以上问题,节省时间、避免污染。
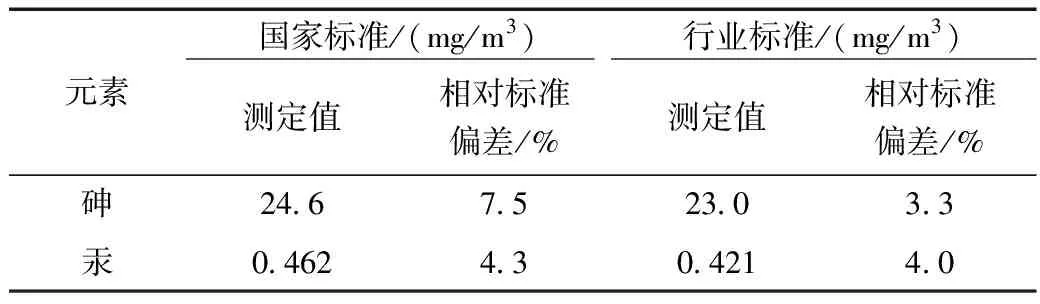
表5 实际样品精密度测试结果
2.3 正确度
为进一步验证两个标准的准确度,在采集的实际样品中,分别取7次加入标准使用液进行加标回收率试验,结果如表6所示,其回收率范围在92%~105%间,均有较好的回收率,其准确度均符合土壤检测要求。
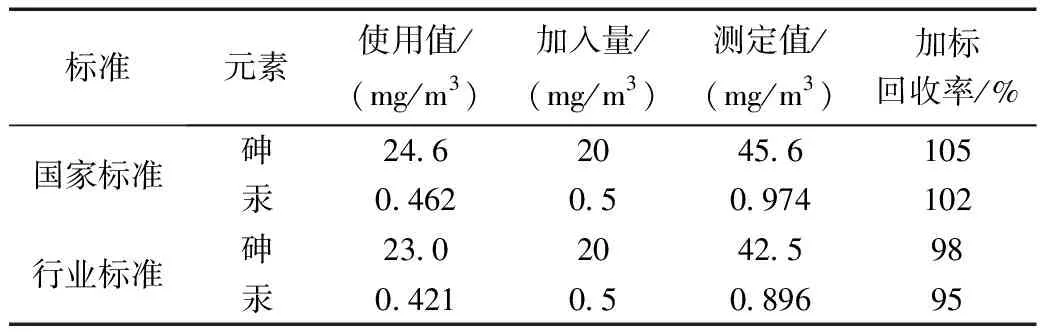
表6 加标回收率测试结果
3 讨 论
3.1 干扰及消除[6]
过渡金属元素如铜等元素高于一定浓度可能会对测定产生干扰,可以加入硫脲-抗血酸溶液可以排除绝大多数干扰。样品中含有100 mg/L以下的Cu2+,50 mg/L以下的Fe3+,1 mg/L以下的Co2+,10 mg/L以下的Pb2+(对硒为5 mg/L),150 mg/L以下的Mn2+(对硒为2 mg/L)均不影响测定。常见的阴离子对测定也无干扰。在排除干扰时,可选用石英管原子化的双层结构,内、外两层均通氩气,在外形成保护层隔绝空气,使待测元素的基态原子在空气中不与氧、氮发生碰撞,减少荧光淬火对测定的作用。
3.2 样品的稀释[7]
测试砷和汞的过程中,通过控制样品溶液的稀释度,是保障测定质量、准确性的关键手段[8]。测定过程中,针对浓度高的超曲线分析溶液,要求工作人员在完成其大致浓度范围的确定后,稀释处理相应的溶液,特别是砷,由于仪器本身缘由使得曲线最高点无法配置太高浓度,否则容易导致仪器受污染降低了灵敏度,因此在上机测试前,可对砷先进行第一步稀释,确保其处于校准曲线测定范围内,这样一来即可为最终获取的数据结果提供准确性以及对仪器的灵敏度提供了保障。
3.3 仪器污染解决方案
3.3.1 先排除器皿污染
取两瓶“娃哈哈”或“屈臣氏”纯净水(不能用矿物质水),例如“娃哈哈”纯净水体积为596 mL,近似为600 mL,用量筒倒去部分水(如一瓶倒去200 mL,一瓶倒去400 mL)。取400 mL瓶,手持优级纯酸瓶直接倒入纯净水瓶中优级纯的酸20 mL,即定量在5%摇匀,作为载流。取200 mL瓶加入1 g的NaOH(或KOH),摇匀溶解后再加入4 g的KBH4(或2.8 g NaBH4)摇匀,作为还原剂。注意固体试剂的称量方法:用新取的滤纸或称量纸,将固体用手在瓶中摇碎,手拍瓶口倒在纸上,称到大致为以上的分量。然后将进样针上的样品进样毛细管拔下和载流管一起直接插入刚配好的400 mL载流瓶中,还原剂管也直接插入刚配好的还原剂娃哈哈瓶中,做空白。 如果污染消除(峰形图上基线笔直、标准空白的荧光值明显降低下来),则刚才是以前配试剂时的玻璃杯、容量瓶、玻璃棒、移液管等器皿的污染。
3.3.2 试剂污染
如果污染没有消除(峰形图基线还是有波峰、爬坡、荧光值很高等),则刚才是酸、氢氧化物、硼氢化物污染(如果刚才使用了玻璃棒来捣碎固体试剂,也有可能是此玻璃棒污染),一样样更换尝试直到找到污染源为止。
3.3.3 室内环境污染(主要是汞污染)
若室内之前有打碎过温度计,或进行过较高浓度的实验,容易引起环境污染。解决方法:一是更换实验室。二是开风扇吹,并在房间中放硫磺(汞污染),隔一段时间再进行尝试。污水厂做氨氮实验的,很容易引起汞污染,坚决避免和氨氮实验相关器皿试剂甚至共用的前处理实验室混用。
3.3.4 仪器污染
进了mg/L 级高浓度的标准溶液,或者基质复杂的土壤、污水样品,拆洗U型反应管和气液分离器,20%的HNO3浸泡24 h,清洗后晾干使用。
4 结 论
选择国家标准《土壤质量 总汞、总砷、总铅的测定原子荧光法》(GB/T 22105-2008)和行业标准《土壤和沉积物 砷、汞、硒、锑、铋的测定 微波消解/原子荧光法》(HJ 680-2013)测定土壤中砷和汞的含量时,选择适宜的仪器工作条件,在前处理和上机测试时,防止受污染。根据测定结果得知,砷和汞的相对标准偏差均处于10%以下,说明两个方法都具备良好的精密度,且加标回收率处于92%~105%范围内,方法准确度较高,适用于土壤中砷汞的测定。因此,在考虑到时间成本和效率问题的情况下,在选用国家标准《土壤质量 总汞、总砷、总铅的测定原子荧光法》(GB/T 22105-2008)的同时,建议把业标准《土壤和沉积物 砷、汞、硒、锑、铋的测定 微波消解/原子荧光法》(HJ 680-2013)一并加进去供工作人员参考并选择。
猜你喜欢
World Journal of Clinical Cases(2020年17期)2020-09-18 08:03:24
机械制造文摘·焊接分册(2020年4期)2020-01-11 01:16:49
检验医学与临床(2020年1期)2020-01-10 04:44:22
中成药(2016年8期)2016-05-17 06:08:22
食品界(2016年4期)2016-02-27 07:37:14
中国医疗器械杂志(2015年5期)2015-12-31 06:15:30
铜业工程(2015年4期)2015-12-29 02:48:44
应用海洋学学报(2015年2期)2015-11-22 07:36:40
中国质量与标准导报(2015年2期)2015-02-28 22:27:16
中国质量与标准导报(2015年2期)2015-02-28 22:27:14
