
肌球蛋白磷酸化靶向亚基家族与心血管疾病
2023-12-13 03:59赵文巧朱柏俊刘宇峰曹明欣邓凯吴心欣吴华英
心血管病学进展 2023年11期
赵文巧 朱柏俊 刘宇峰 曹明欣 邓凯 吴心欣 吴华英
(湖南师范大学医学院,湖南 长沙 410013)
心血管疾病是全世界成人死亡的主要原因[1]。血管平滑肌功能失调是多种心血管疾病发生发展的共同特点。肌球蛋白磷酸化靶向亚基(myosin phosphatase targeting subunit,MYPT)作为肌球蛋白轻链磷酸酶(myosin light chain phosphatase,MLCP)的靶向亚基,可通过磷酸化调控MLCP活性而影响肌球蛋白调节轻链(myosin regulatory light chain,MLC)的磷酸化,影响细胞功能,参与对血管平滑肌收缩与舒张过程的调节[2]。已有研究表明MYPT家族参与多种心血管疾病的发生发展,如心脏肥大、心力衰竭、原发性高血压、冠心病、脑卒中等。了解MYPT家族在心血管疾病中的研究现状,有助于阐明疾病的发生机制,并为其治疗提供新思路。
1 MYPT家族成员结构
MYPT家族包括5个成员,分别是MYPT1(PPP1R12A)、MYPT2(PPP1R12B)、MYPT3(PPP1R16A)、MBS85(PPP1R12C)、TIMAP(PPP1R16B)。有研究[3-4]比较了MYPT家族5个成员的序列,发现MYPT家族成员共享几个保守结构域,包括位于N端附近用于与蛋白磷酸酶1催化亚基(protein phosphatase 1 catalytic subunit,PP1c)结合的RVxF基序,以及紧随其后的几个锚蛋白重复序列。锚蛋白重复序列形成了一个与多种蛋白质结合的交互平台,包括底物磷酸化肌球蛋白。此外,还发现MYPT1、MYPT2和MBS85含有C-末端亮氨酸拉链(leucine zipper,LZ)结构域,LZ结构域在NO/cGMP/PKG通路对MLCP活性的调控中发挥重要作用[5]。MYPT3和TIMAP缺乏LZ结构域,但它们含有1个C-末端CAAX盒,可将蛋白靶向到质膜上[3]。MYPT家族成员受各种蛋白激酶在多个位点的磷酸化调节。研究发现Rho相关激酶(Rho-related kinase,ROCK)通过催化Thr696和Thr853位点的磷酸化来调节MYPT1[6],这两个位点在MYPT2中是保守的,而在MBS85中只有Thr696对应的位点保守,在TIMAP和MYPT3中则没有发现这两个位点[3]。
尽管MYPT家族成员之间有很多相似性,但它们的组织分布却有着一定的差异。MYPT1主要在平滑肌中表达,而MYPT2主要在脑和横纹肌中表达。MBS85是一种普遍表达的蛋白,它是强直性肌营养不良相关Cdc42结合激酶-α的底物,介导了由Cdc42诱导的肌动蛋白重组[7]。MYPT3在心脏、大脑和肾等多个器官中表达,与其他成员不同的是,MYPT3与PP1c结合会抑制后者对MLC的催化活性[8]。TIMAP是MYPT家族的内皮特异性成员,主要定位于内皮细胞的质膜上,并通过与非整合蛋白层粘连蛋白受体1和埃兹蛋白/根蛋白/膜突蛋白(ezrin/radixin/moesin proteins,ERM蛋白)等的相互作用发挥广泛的生理功能[9]。
2 MYPT家族成员与细胞功能
2.1 细胞收缩
细胞收缩装置主要由肌动蛋白和肌球蛋白组成,此装置既依赖于Ca2+又依赖于磷酸化,基础Ca2+水平或MLC磷酸化状态的恢复会使收缩的肌肉恢复到非收缩状态。平滑肌收缩取决于细胞质内Ca2+浓度的增加,Ca2+可与钙调蛋白结合,进而激活钙调蛋白依赖性肌球蛋白轻链激酶(myosin light chain kinase,MLCK),使其磷酸化MLC的Ser19或Thr18,磷酸化的MLC与肌动蛋白相互作用,从而导致平滑肌收缩[10]。横纹肌收缩也涉及此机制,但不是其主导机制[10]。
MLC去磷酸化由MLCP介导。MLCP是一个异源三聚体,由PP1c、MYPT和M20组成[11]。PP1c是一种丝氨酸/苏氨酸磷酸酶,是MLCP发挥作用的核心酶。MYPT通过RVxF基序与PP1c结合,并将其特异性导向MLC,从而赋予MLCP特异性[2]。MYPT的磷酸化会影响PP1c与MLC结合,从而调节MLC磷酸化水平,改变平滑肌舒缩状态。
2.2 细胞运动
细胞运动中最关键且必需的事件之一是MLC的循环磷酸化和激活[12]。MLCK或ROCK催化MLC磷酸化,从而激活肌球蛋白,使肌球蛋白能结合肌动蛋白并利用ATP产生运动。相反,MLCP对MLC的去磷酸化则降低了肌球蛋白对肌动蛋白的亲和力。MYPT通过调节MLCP活性影响MLC的磷酸化,从而参与调控细胞运动过程。
2.3 胞质分裂
收缩环是胞质分裂的重要结构,主要由肌动蛋白和肌球蛋白Ⅱ装配而成。有研究[13]通过比较过表达不可磷酸化的MLC(T18A/S19A)细胞和野生型MLC细胞,发现前者收缩环排列受阻,胞质分裂不完全,而野生型MLC细胞收缩环排列和胞质分裂均正常。这揭示MLC Thr18/Ser19的磷酸化对收缩环的正常排列是重要的。MYPT对MLCP活性的调节可影响MLC磷酸化,从而参与调控胞质分裂过程。
2.4 细胞骨架
细胞骨架参与细胞分裂和收缩等多种细胞功能。有研究[14]通过测定一个网柄菌肌球蛋白缺陷突变体的细胞骨架组织和生理反应,发现其微管网络形态和分布异常,且细胞分裂等功能也存在异常。由此推测肌球蛋白可能与细胞骨架存在某种联系。随后有研究[15]发现肌球蛋白Ⅱ参与细胞骨架的组装,并在细胞骨架中与F-肌动蛋白紧密结合,并通过MLC Thr18/Ser19的磷酸化来调节其功能。MYPT可影响MLC的磷酸化,从而参与调控细胞骨架的组装和功能。
ERM蛋白为跨膜蛋白和底层细胞骨架之间的接头,特殊位点的磷酸化可使其活化而发挥功能[16]。研究[17]发现MLCP可通过介导ERM蛋白的去磷酸化在细胞骨架中发挥调节作用。此外,MYPT与微管相关蛋白Tau和微管相关蛋白2存在相互作用关系,提示MYPT可能调节微管动力学[18]。
2.5 细胞黏附
研究[19]发现,p-MYPT/MYPT比值的增加会促进单核细胞-内皮细胞黏附,这表明MYPT可通过磷酸化调控单核细胞-内皮细胞黏附。此外,肌动蛋白细胞骨架可调节钙黏蛋白之间的相互作用,后者在调节细胞-细胞黏附中发挥重要作用[20]。肌球蛋白对肌动蛋白细胞骨架至关重要,MYPT通过调节MLC的磷酸化而影响其功能,从而影响肌动蛋白细胞骨架的正常形成及功能,并借此调节钙黏蛋白的相互作用而影响细胞-细胞黏附。
3 MYPT家族成员与信号通路
3.1 Rho/ROCK信号通路
Rho家族是鸟嘌呤核苷酸结合蛋白RAS超家族的组成部分,RhoA是该家族中研究最多的一个。RhoA及其下游靶标ROCK在细胞收缩、迁移等广泛的生理过程和Rho/ROCK信号通路中发挥重要作用[21]。ROCK是一种丝氨酸/苏氨酸激酶,有两种亚型,ROCK1和ROCK2[22]。它们在体内普遍表达,ROCK1在肝、脾、肾等非神经性组织中表达水平较高,而ROCK2主要在脑、肌肉和心脏中表达[23]。RhoA/ROCK通路的上游信号之一是血管紧张素Ⅱ或其他多肽,如内皮素-1、成纤维细胞生长因子和转化生长因子-β与细胞膜上的受体结合[24],然后通过其受体G偶联蛋白来动员Gq蛋白的G-α亚单位(Gαq),从而激活Rho-鸟嘌呤交换因子,以鸟苷三磷酸(guanosine triphosphate,GTP)取代鸟苷二磷酸[25]。随后GTP-Rho通过与ROCK结构中的Rho结合结构域相互作用使ROCK激活[26]。
ROCK可磷酸化MYPT1,抑制MLCP的活性,使MLC持续磷酸化,从而参与调控肌球蛋白基础上的细胞收缩、黏附等一系列细胞功能[27]。研究发现Rho/ROCK通路在许多病理生理过程中发挥重要作用,如心力衰竭[28]和动脉粥样硬化[29]等。
3.2 NO/cGMP/PKG通路
一氧化氮(nitric oxide,NO)是重要的血管内皮舒张因子,其舒张血管作用主要是通过增加环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)的产生,进而激活cGMP依赖性蛋白激酶G(protein kinase G,PKG)而实现。PKG如何实现对血管的调控?研究[30]发现,PKG通过与MYPT1结合介导MLCP活化,从而导致Ca2+脱敏和血管舒张。MYPT1亚基可通过3’端31bp外显子的选择性剪接,生成不同的C-末端LZ阳性(LZ+)或LZ阴性(LZ-)结构域[31]。NO/cGMP/PKG通路诱导的血管舒张与MYPT-LZ+/LZ-有关。当缺乏LZ结构域时,PKG可与MYPT1-LZ-结合,但磷酸化和Ca2+脱敏不会发生。当LZ表达时,NO下游的cGMP、PKG和MLCP活性增加[5]。这种效应需要PKG1α磷酸化MYPT1[30]。PKG1α在MYPT1上的磷酸化位点主要是Ser695,此位点的磷酸化会干扰相邻Thr696的磷酸化,从而消除磷酸化Thr696对MLCP的抑制,重新激活MLCP并导致Ca2+脱敏,进而诱导平滑肌松弛[32](见图1)。
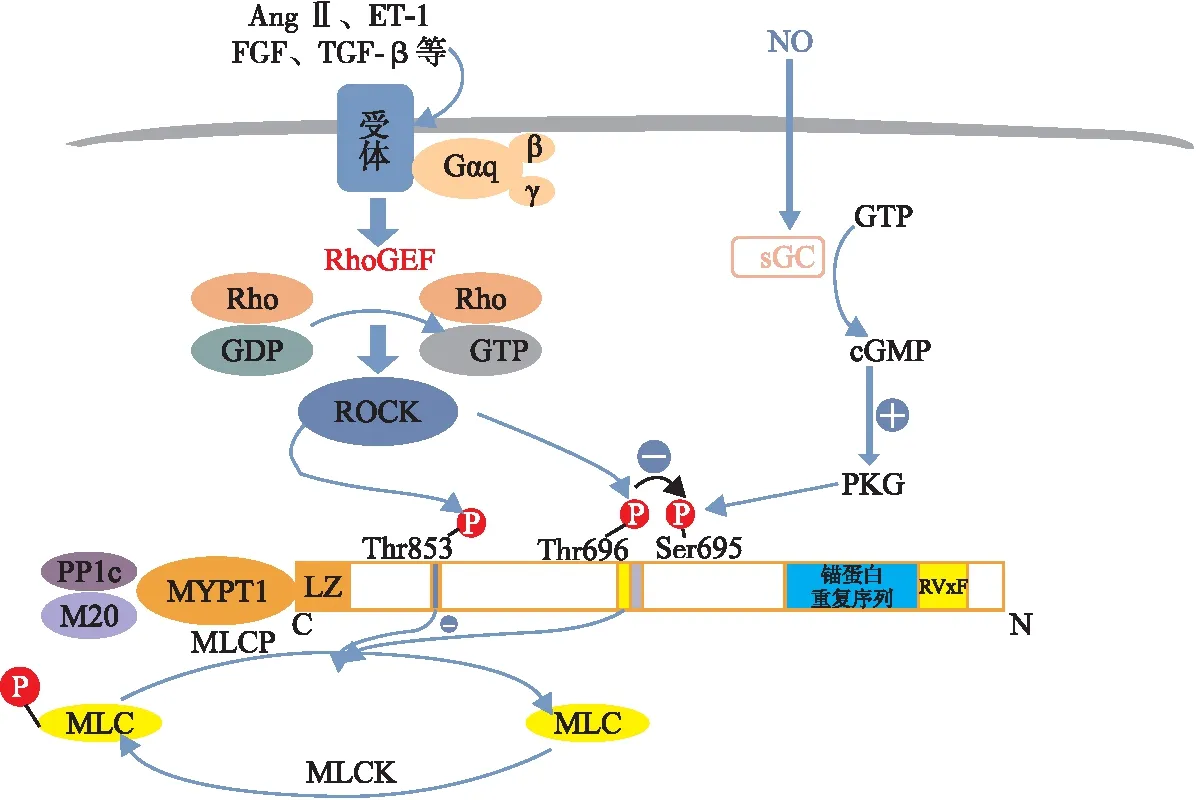
注:AngⅡ,血管紧张素Ⅱ;ET-1,内皮素-1;FGF,成纤维细胞生长因子;TGF-β,转化生长因子-β;RhoGEF,Rho-鸟嘌呤交换因子;GDP,鸟苷二磷酸;sGC,可溶性鸟苷酸环化酶;PKG,蛋白激酶G。
4 MYPT家族成员与心血管疾病
4.1 心脏肥大
心脏肥大多由主动脉狭窄和高血压等应激引起,最初是一种保护性的代偿机制,但持续和过度应激诱导的病理性心脏肥大是不利的,并最终可能导致心力衰竭等严重后果。
研究[33]表明MLC磷酸化可抑制心脏肥大。MLCK家族由MLCK-1、MLCK-2、MLCK-3和MLCK-4组成[34]。心肌MLC的磷酸化主要由MLCK-3即cMLCK调控,cMLCK在心房和心室中高度且等水平表达,且对培养物中分离的心肌细胞发挥正性肌力作用[35]。此外,MLCK-4也在心肌中表达[36]。故心肌MLC磷酸化可由两种蛋白激酶来调节。
在心脏中,MYPT1和MYPT2都表达,但MYPT2比MYPT1表达更丰富[37]。MYPT2可调节MLCP活性,影响MLC的去磷酸化,从而调控心肌MLC的磷酸化水平,影响心脏肥大的发生发展。
4.2 心力衰竭
心力衰竭是由心排血量减少不能满足机体需要而导致的一种临床综合征。研究[38]表明心肌MLC磷酸化的变化在心力衰竭进展过程中发挥重要作用。MYPT2作为心脏MLCP的靶向亚基,可影响心肌MLC去磷酸化从而参与对心力衰竭的调控。
充血性心力衰竭是心力衰竭的一种常见类型,其特征是血管收缩异常和对NO介导的血管舒张效应受损[39]。MYPT1-LZ+亚型是NO发挥血管舒张效应的重要结构[3]。研究[39]表明,心力衰竭患者对NO介导的血管舒张敏感性降低可部分归因于MYPT1-LZ+亚型表达水平的降低。故临床上可使用调节MYPT-LZ+亚型表达的药物,如血管紧张素转化酶抑制剂在治疗心力衰竭上效果较好[3]。
4.3 高血压
高血压是最常见的心血管疾病,也是导致脑卒中、冠心病等疾病的重要危险因素。高血压的一个重要特征是血管阻力增加,血管收缩增强。MYPT可影响MLC的磷酸化水平,从而调控血管舒缩。
平滑肌的主要MYPT亚型是MYPT1。ROCK可磷酸化MYPT1的Thr696、Thr853位点而抑制MLCP活性,使MLC磷酸化水平增高。此外,ROCK还可使蛋白激酶C磷酸酶抑制蛋白17 (C-kinase-potentiated protein phosphatase 1 inhibitor of 17 kDa,CPI-17)磷酸化[40],磷酸化的CPI-17通过与PP1c结合来抑制MLCP活性[41]。使用Y27632(ROCK抑制剂)阻断ROCK活性对多种实验性高血压大鼠均具有降血压作用[42]。阻断ROCK活性可能成为高血压治疗的重要靶点。
4.4 冠心病
冠状动脉痉挛在多种缺血性心脏病的发病机制中起着重要作用,而痉挛的中心机制是血管平滑肌的过度收缩[43]。MYPT1可通过影响MLC磷酸化而参与调节血管平滑肌舒缩状态,在冠状动脉痉挛中发挥重要作用。
急性冠脉综合征是冠心病的一种严重类型,多由动脉粥样硬化斑块破裂伴血栓形成引起。研究[44]表明,血小板活化并形成血栓是急性冠脉综合征发病的一个重要机制。血小板形态变化是血小板活化的早期事件,MLC磷酸化以及与细胞骨架结合是触发血小板形态变化的主要原因[45]。血小板中的MYPT亚型主要是MYPT1。MYPT1可影响MLC的磷酸化,调控血小板活化。
Rho/ROCK通路在血小板活化过程中发挥重要作用[46],此外,还参与内皮功能障碍的调节、炎症等,在动脉粥样硬化的发生发展中发挥重要作用[29]。另有研究[47]发现,ROCK的抑制显著限制了喂食无胆酸盐高脂肪饮食的低密度脂蛋白受体敲除小鼠的早期动脉粥样硬化斑块的发展。这表明抑制ROCK是治疗动脉粥样硬化的潜在靶点。
4.5 脑卒中
缺血性脑卒中由大脑缺血缺氧而引起。小胶质细胞和T细胞等炎症细胞介导的神经炎症在缺血性脑卒中的发病机制中起重要作用[48]。研究发现,ROCK抑制剂Fasudil可抑制小胶质细胞分泌促炎因子[49],且ROCK在T细胞的活化及跨内皮迁移等过程中发挥重要作用[50]。此外,血小板ROCK2对血栓形成和稳定至关重要,是血栓栓塞性脑卒中的重要致病介质[51]。MYPT作为Rho/ROCK通路的主要下游靶标,参与T细胞跨内皮迁移[50]、血小板活化等多个过程的调控,在脑卒中的发病过程中发挥重要作用。
5 结语
MYPT家族成员在结构上存在相似性,但在组织分布上具有差异,MYPT对MLC磷酸化在参与细胞收缩、细胞运动、细胞黏附等生物学过程中发挥重要作用,参与Rho/ROCK通路与NO/cGMP/PKG通路的调控。MYPT1作为ROCK的主要下游靶标,是目前MYPT家族研究最为广泛的,参与许多心血管疾病如心脏肥大、心力衰竭及高血压等的发生发展,家族其他成员如TIMAP与心血管疾病的联系也有报道,但家族成员之间是否存在相互作用影响疾病的发生发展值得探讨。总之,MYPT家族在心血管疾病中的功能表现,有望成为疾病防治的潜在靶标。
猜你喜欢
食品科学(2023年4期)2023-03-06
世界科学技术-中医药现代化(2022年3期)2022-08-22
中国畜牧杂志(2020年1期)2020-01-16
生物学通报(2019年3期)2019-06-15
浙江农业科学(2016年11期)2016-05-04
中国学术期刊文摘(2016年2期)2016-02-13
中国医科大学学报(2015年10期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
心血管病学进展(2015年4期)2015-02-22
中国医学科学院学报(2013年6期)2013-03-11
