
黑色素瘤缺乏因子2炎症小体在心血管疾病中的研究进展
2023-12-13 03:59:34韩冰来春林
心血管病学进展 2023年11期
韩冰 来春林
(山西医科大学第五临床医学院心血管内科,山西 太原 030012)
心血管疾病(cardiovascular disease,CVD)是一类严重威胁人类健康和生命安全的重大疾病,据《中国心血管健康与疾病报告2021》统计表明,CVD发病率及死亡率居高不下,且发病人群的年龄逐渐下降,已成为当今社会需面对的一项严重的公共卫生问题。
动脉粥样硬化(atherosclerosis,AS)是造成CVD重要的病理生理基础,目前认为该病是一种由脂质驱动的慢性炎症性疾病[1],其典型病理表现为血管内膜形成粥瘤或纤维斑块,造成血管管腔狭窄及内膜损伤,最终导致CVD的发生。AS形成机制经历了多种学说,目前炎症学说受到广泛关注,认为炎症贯穿了AS的发生全过程并对其预后也有一定影响,炎症小体在炎症发生过程中起到重要作用。最近多项研究表明黑色素瘤缺乏因子2(absent in melanoma 2,AIM2)参与了多种CVD的发生和发展过程,现就AIM2炎症小体在CVD中的最新研究进展进行综述。
1 AIM2生物学功能概述
AIM2是一种细胞质传感器,可特异性识别病原体释放入胞浆的双链DNA。Kumari等[2]首先发现AIM2基因位于1号染色体长臂(q22),与造血干扰素诱导核蛋白(hematopoietic interferon-inducible nuclear protein domain proteins,HIN200)家族成员干扰素诱导蛋白16和髓样细胞核分化抗原有保守序列,认为其属于免疫相关的干扰素诱导p200蛋白家族中的一员。从结构上看,AIM2包含N端热蛋白结构域(pyrin-like domain,PYD)和C端具有200个氨基酸重复序列的HIN200结构域[3],HIN200结构域包含寡核苷酸/寡糖结合折叠的亚域,可与蛋白质、寡糖或核苷酸结合。PYD结构域属于进化上高度保守的死亡结构域超家族中的一员,可参与蛋白质相互作用,多存在于与细胞凋亡和炎症相关的蛋白质中[4]。
炎症小体是一种多聚体蛋白复合物,其主要组成部分有传感器、包含胱天蛋白酶-1(caspase-1)募集结构域(caspase recruitment domain,CARD)的凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a CARD,ASC)和caspase-1前体蛋白(pro-caspase-1)。当识别特定刺激后,传感器AIM2、NOD样受体热蛋白结构域相关蛋白3(NOD-like receptor pyrin domain containing 3,NLRP3)、核苷酸结合寡聚化结构域样受体蛋白(NOD-like receptor family pyrin domain-containing protein 4,NLRC4)等与ASC结合,促进pro-caspase-1聚集,促进体内炎症小体的组装及其介导的炎症反应[5]。当宿主被感染后,AIM2的HIN结构域可识别并结合释放到胞浆中的病原体双链DNA,随后PYD与HIN结构域解离,并通过AIM2PYD-AIM2PYD相互作用招募并结合ASC,形成AIM2-ASC复合物。ASC是炎症小体中常见的连接蛋白,ASC可参与ASCCARD-Casp1CARD的相互作用,招募具有同样CARD结构域的pro-caspase-1,pro-caspase-1通过二聚化和自体蛋白水解,生成p10和p20亚基,2个p10亚基与2个p20亚基结合形成具有蛋白水解活性的caspase-1,最终形成炎症复合体(AIM2-ASC-pro-caspase-1)[4]。活化的caspase-1可剪切前白细胞介素(pro-interleukin,pro-IL)-1β和pro-IL-18,介导白细胞介素(interleukin,IL)-1β和IL-18的成熟和分泌。此外,caspase-1可将消皮素D(gasdermin D,GSDMD)切割为N端和C端,GSDMD-N会造成胞质膜穿孔,破坏细胞膜完整性,造成细胞肿胀、裂解和死亡,并释放IL-1β和IL-18等炎症因子(如图1)。IL-1β介导的炎症反应在多种CVD中发挥重要作用[6]。此外,近期多项研究[7]证明由caspase-1介导的细胞焦亡在CVD的发展过程中也占据了重要地位。
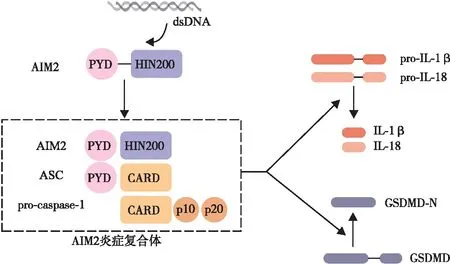
注:dsDNA,双链DNA。
2 AIM2与CVD
2.1 AIM2与动脉粥样硬化性心血管疾病
动脉粥样硬化性心血管疾病是威胁中国居民健康的重要疾病,AS是造成动脉粥样硬化性心血管疾病的病理基础,现认为内皮细胞损伤、炎症反应、脂质代谢异常、血流动力学改变均对其有一定影响。近年来多项实验研究[8]证明,AS是一种脂质驱动的慢性进行性炎症性疾病。在AS发病早期,内皮细胞在其表面选择性地表达黏附分子,使多种类型的白细胞发生黏附并向内皮损伤处迁移。单核细胞趋化蛋白-1可刺激单核细胞转化为巨噬细胞,促进低密度脂蛋白氧化修饰为氧化低密度脂蛋白,氧化低密度脂蛋白会与单核/巨噬细胞的清道夫受体结合形成泡沫细胞,同时生成活性氧自由基、蛋白酶、脂肪酶,并高表达清道夫受体和Toll样受体。此外,巨噬细胞和受损的内皮细胞还会释放IL-6、IL-8和单核细胞趋化蛋白-1等因子,引发进一步炎症级联反应。在AS的进展期,血管内皮细胞及血管平滑肌细胞会生成基质金属蛋白酶,降解细胞外基质的胶原纤维,造成AS斑块破裂、出血以及血栓形成[9]。最近多项研究证明AIM2在AS的发展过程中起着多重作用。
2.1.1 AIM2与内皮功能障碍
血管内皮细胞功能障碍是AS发生的起始环节,首先血液中的白细胞会向血管内皮损伤处迁移和黏附,受损的内皮细胞也会继续释放炎症因子,诱发炎症反应的级联放大[10]。血管内皮细胞可分泌一氧化氮、内皮素-1、前列腺素、血栓素A2、血管紧张素Ⅱ、细胞间黏附分子-1、血管细胞黏附分子-1等多种物质,参与血管紧张性调节、血管纤维蛋白溶解、血管平滑肌细胞增殖调控、炎症细胞因子黏附和迁移等进程[11]。
poly(dA:dT)(双链DNA,AIM2特异性刺激因子)、炎症因子如肿瘤坏死因子-α和γ干扰素可增加内皮细胞AIM2的表达。Lüsebrink等[12]研究发现,在ApoE-/-的小鼠中,给予持续皮下注射poly(dA:dT)的小鼠,其细胞活性氧及循环内皮微粒的数量增加,造成内皮功能进一步紊乱,加重冠状动脉炎症反应,增加AS斑块形成。在AIM2-/-的颈动脉损伤小鼠模型中发现,与对照组相比,使用poly(dA:dT)不会破坏血管再内皮化。综上所述,刺激AIM2炎症小体可通过促进内皮细胞迁移和凋亡,破坏再内皮化进程,促进AS的发展及AS斑块的形成。此外,Pan等[13]研究发现AIM2炎症小体可增加细胞间黏附分子-1的表达,细胞间黏附分子-1可与淋巴细胞功能相关抗原-1结合,促进单核细胞黏附于内皮细胞,促进泡沫细胞形成,这可能是AIM2炎症小体诱导AS形成的机制之一。
2.1.2 AIM2与巨噬细胞
潜能未定克隆性造血(clonal hematopoiesis of indeterminate potential,CHIP)的主要特征病变为髓系肿瘤相关基因体细胞突变。目前有多项研究[14]证明,携带CHIP与AS的发生和发展及心脑血管疾病风险增加有关。CHIP的发病率随着年龄的增长而升高,这是由于表观遗传修饰基因DNMT3A、TET2、ASXL1和酪氨酸激酶基因JAK2发生突变所致。JAK2功能获得性突变为JAK2VF,其与骨髓增生性肿瘤和动脉粥样硬化性心血管疾病的发病风险增加有关。一些研究表明,巨噬细胞是连接AS与CHIP的重要细胞。JAK2VF突变会使细胞代谢发生变化,造成DNA氧化受损,激活AIM2炎症小体,促进IL-1β的释放,加速巨噬细胞的增殖。AIM2炎症小体激活也会促进细胞焦亡的进程,形成前馈环路,对巨噬细胞增殖和细胞焦亡有双重影响[15]。在AS病变中,巨噬细胞中的AIM2炎症小体激活可形成坏死核并促进疾病发展。此外,AIM2炎症小体可能参与了巨噬细胞的胆固醇代谢过程。AIM2炎症小体激活caspase-1,切割GSDMD产生GSDMD-N端。GSDMD不光参与IL-1β、IL-18的释放及细胞焦亡的过程,也可通过促进胆固醇外排来促进泡沫细胞的形成[16]。
2.1.3 AIM2与血管平滑肌细胞
血管平滑肌细胞(vascular smooth muscle cells,VSMCs)是动脉中膜的重要组成部分,VSMCs迁移入内膜并增生,是AS进展期病变形成的重要环节。在AS进展期,VSMCs可产生基质金属蛋白酶,而基质金属蛋白酶可调节细胞外基质,与动脉炎症反应密切相关,目前被认为是评估AS的炎性指标之一。促炎因子肿瘤坏死因子-α和γ干扰素可激活VSMCs中的AIM2炎症小体[17]。AIM2的激活又会增强VSMCs中转化生长因子-β、果蝇母性DPP同源蛋白2和果蝇母性DPP同源蛋白3的活性,促进AS斑块形成过程中基质金属蛋白酶的表达,促进VSMCs的迁移[18]。氧化低密度脂蛋白可通过促进活性氧的生成上调VSMCs中的AIM2/ASC/caspase-1通路和GSDMD-N,从而促进VSMCs焦亡[19](如图2)。
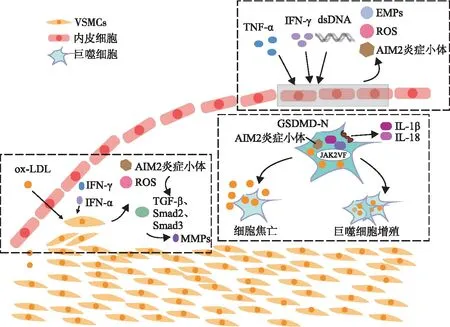
注:ox-LDL,氧化低密度脂蛋白;IFN-γ,γ干扰素;IFN-α,α干扰素;TNF-α,肿瘤坏死因子-α;ROS,活性氧;Smad,果蝇母性DPP同源蛋白;MMPs,基质金属蛋白酶;TGF-β,转化生长因子-β;EMPs,循环内皮微粒;dsDNA,双链DNA。
2.1.4 AIM2与脂质代谢
除了斑块局部作用外,AIM2炎症小体对脂质代谢也有一定影响,进一步加重AS的进程,同样,多种脂类物质也参与调节AIM2的表达。高脂饮食会增加ApoE-/-小鼠动脉中的AIM2、细胞间黏附分子-1和GSDMD-N的表达。高密度脂蛋白会降低尼日利亚菌素、胆固醇结晶、NLRP3炎症小体激活剂、AIM2炎症小体激活剂以及Toll样受体激动剂的促炎作用,减少IL-6和IL-1β的生成[20]。有研究发现,AIM2-/-小鼠会在白色脂肪组织诱导炎症和脂肪生成,同时造成葡萄糖稳态受损,最终导致肥胖和胰岛素抵抗。此外,AIM2-/-小鼠能量消耗减少,并出现棕色脂肪组织功能受损。这些证据表明AIM2在能量代谢、炎症和胰岛素抵抗中起着重要作用[21]。虽然这项研究表示AIM2可以预防肥胖和胰岛素抵抗,但是AIM2也以其他方式参与并促进了AS斑块形成的进程。因此,AIM2炎症小体在AS中的作用仍有待进一步研究。
2.2 AIM2与心力衰竭
心力衰竭(心衰)是一种由于心脏收缩或舒张功能障碍引起的临床综合征,其不能满足机体正常代谢需要。心衰发病率与年龄显著相关,随着年龄增长而升高,在65岁以上人群中发病率占4%~8%。目前认为,心衰的发生与慢性炎症有一定关系,疾病发生时其炎性因子如C反应蛋白、肿瘤坏死因子、IL-1、IL-6、IL-18等均有明显增加[22],同时对患者预后也有一定预测价值。
糖尿病是CVD发生的高危因素,其通过多种机制增加心肌对缺血再灌注损伤的易感性[23],升高心衰的发病率,炎症反应在其中发挥主要作用。Durga Devi等[24]研究认为,糖尿病小鼠心肌梗死后出现左心室功能不全,梗死区域及其周围的心肌细胞出现线粒体自噬功能障碍,线粒体DNA释放增加,激活AIM2炎症小体、NLRC4炎症小体及IL-18,造成caspase-1过度活化,同时使用免疫荧光染色和流式细胞术分析显示,糖尿病小鼠左心室中M2型巨噬细胞减少,而促炎型M1型巨噬细胞增加,也可认为AIM2炎症因子通过启动炎症反应参与了心肌梗死和心衰的过程。在高糖处理的H9c2细胞中,AIM2可调节GSDMD-N通路的表达,影响心肌细胞焦亡和心室重塑,影响心功能[25]。
AIM2炎症小体的激活已被证明在糖尿病小鼠梗死后的早期心衰中起着重要作用。Onódi等[26]为进一步研究AIM2炎症小体在缺血再灌注损伤诱发的慢性心衰晚期的激活,分别从3个时间节点对缺血再灌注损伤猪模型的左心室组织进行评估,发现心脏组织中AIM2炎症小体的水平在3 h及3 d时无明显改变,但在2个月时明显升高。
2.3 AIM2与心肌炎
心肌炎和炎症性心肌病是造成心衰发生的常见疾病,也是CVD中的难点。心肌炎的特点是心肌出现局限性或弥漫性的炎性病变。Toldo等[27]在急性病毒性心肌炎患者的心脏活检样本中,发现ASC及caspase-1的胞内聚集物,认为急性心肌炎患者心脏中存在以炎症小体形成为特征的组织炎症反应。B3型柯萨奇病毒是造成心肌炎发生的主要病因之一,会导致急性或慢性病毒性心肌炎,病情加重时会发展为扩张型心肌病。
一项临床研究认为在心肌炎和扩张型心肌病患者的心肌组织中存在肠病毒衣壳蛋白[28],肠道病毒可能产生病毒蛋白和RNA,造成心脏持续感染,促进病毒性心肌炎发展为扩张型心肌病。在B3型柯萨奇病毒诱导的慢性心肌炎动物模型中,分别接种壳聚糖-pVP1(CS-pVP1)、壳聚糖-pAIM2(CS-pAIM2)、CS-pAIM2联合CS-pVP1,发现CS-pAIM2/CS-pVP1联合免疫会促进CD8+记忆T细胞和CD8+多功能T细胞的诱导来缓解B3型柯萨奇病毒引起的慢性心肌炎[29]。此外,研究还发现CS-pAIM2也可减少心肌损伤和纤维化,改善心功能。根据这些结果推测,AIM2炎症小体可能是治疗急性或慢性心肌炎的一种新方法。但AIM2在心肌炎中的确切作用及病理机制仍不清楚,需进一步的研究。
3 以AIM2为靶点的药物治疗
2016年CANTOS研究[30]证明了IL-1β是AS抗炎治疗的有效靶点。COLCOT研究[31]证实秋水仙碱可部分抑制NLRP3炎症小体的形成。以上结果证实了IL-1β靶点的临床重要性及其与CVD治疗的相关性。当AIM2炎症小体组装后,活化的AIM2炎症小体具有caspase-1的蛋白水解活性,可剪切pro-IL-1β和pro-IL-18,介导IL-1β和IL-18的成熟和分泌。在AS、心肌梗死、主动脉瘤、心肌缺血再灌注损伤和心衰中起重要作用[7]。近年来AIM2炎症小体的调控机制广受关注,目前发现了一些可抑制AIM2的药物,为进一步开发AIM2靶向抑制剂提供了一些帮助。
3.1 潜在的AIM2抑制剂
槲皮素是一种具有多种生物活性功能的黄酮类化合物,具有抗氧化、抗炎、免疫调节等作用,其可抑制poly(dA:dT)诱导的IL-18、IL-1β的分泌,主要是通过刺激γ干扰素抑制JAK2/STAT1通路,抑制AIM2和pro-caspase-1的表达[32]。
组蛋白去酰化酶3是一种蛋白酶,主要功能为染色体结构的修饰及基因表达的调控。最新研究[33]发现,长链非编码RNA牛磺酸上调基因1与miR-132-3p结合,上调组蛋白去酰化酶3的表达,抑制抗氧化基因Prdx2、Hsp70及Bcl-xL的表达,介导心肌细胞发生氧化损伤。研究发现,RGFP966作为一种HDAC3的抑制剂,可抑制AIM2炎症小体及HDAC3的表达,并减轻氧化应激反应及其造成的心肌损伤。其发生的可能机制为转录激活因子STAT1的乙酰化和磷酸化、对信号转导的调节作用及对IL-1β的抑制作用。此外,TRIM11被发现有抑制AIM2炎症小体的功能,并推测其可能是通过促进p62介导的选择性自噬途径来降解AIM2[34]。而另一项研究[35]表明自噬抑制剂3-甲基腺嘌呤可抑制RGFP966对AIM2炎症小体的抑制作用,认为RGFP966与AIM2之间存在潜在的自噬调节关系。
3.2 以AIM2炎症小体为靶点的调控剂
有研究[18]认为来源于端粒结构的抑制性寡核苷酸A151,可选择性降低AIM2炎症小体和环鸟苷-腺苷酸合成酶的表达,对AS有一定的保护作用。此外,17β-雌二醇和孕酮可减轻缺血性脑卒中诱导的大鼠AIM2炎症小体和NLRC4炎症小体的上调[36]。
虽然目前AIM2炎症小体抑制剂还未应用于CVD的临床治疗中,但AIM2作为治疗CVD的靶点的有效性已在多种动物实验中得到了证实。AIM2炎症小体抑制剂对于CVD的机制及作用处于初步探索阶段,未来还需在更多的动物实验和临床研究中证明这一观点。
4 小结
在与CVD发病相关的炎症小体中,NLRP3炎症小体受到广泛关注,但是近年来多项研究证明AIM2炎症小体在CVD的发生和发展过程中也起到了重要作用,且AIM2在不同类型的疾病中的表达及作用也不尽相同。综上所述,AIM2可能是治疗CVD的有效干预靶点,但关于AIM2炎症小体抑制剂对于CVD的机制及作用处于初步探索阶段,缺乏有效的临床证据,有待进一步研究。未来的研究方向应更多关注于CVD中AIM2炎症小体的调控机制及信号通路,这将有助于发现新的疾病干预靶点,对CVD的治疗至关重要。
猜你喜欢
祝您健康(2024年1期)2024-01-11 04:39:32
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:43
长春中医药大学学报(2017年1期)2017-04-16 05:56:51
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
中国继续医学教育(2015年4期)2016-01-07 07:38:10
西南军医(2015年3期)2015-04-23 07:28:32
国际心血管病杂志(2015年5期)2015-02-27 12:11:34
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
