
高氧暴露BPD 小鼠模型中巨噬细胞焦亡的检测及意义
2023-12-13 02:50涂子坤高雅静郭海艳周玉峰
复旦学报(医学版) 2023年6期
涂子坤 高雅静,3 韩 晓 郭海艳 周玉峰,3△
(1国家儿童医学中心/复旦大学附属儿科医院儿科研究院 上海 201102; 2国家卫生健康委员会新生儿疾病重点实验室(复旦大学) 上海 201102; 3复旦大学生物医学研究院 上海 200032)
支 气 管 肺 发 育 不 良(bronchopulmonary dysplasia,BPD)是一种早产儿常见的慢性肺病,是早产儿最危重的并发症之一。BPD 临床表现为呼吸严重困难,致使早产儿的预后不良和生存质量下降。随着出生后外源性表面活性物质、产前产后皮质类固醇和早期无创通气等在临床应用,早产儿的存活率上升,但BPD 的发病率却逐年上升[1-2]。BPD 不仅严重影响患儿的身心健康,也给家庭和社会带来沉重的负担。在美国,治疗BPD 的成本约为30 亿美元/年,且重症BPD 合并肺动脉高压的病死率可高达50%[3-4]。目前临床上缺乏治疗BPD 的有效措施,需要进行综合治疗和管理[5]。故积极探索BPD 的发病机制和防治策略,改善患儿的生存质量,具有重要的社会意义和研究价值。
BPD 的发病机制比较复杂,主要是在基因易感性的基础上,各种产前和出生后刺激因素损害脆弱的肺组织,如高氧、机械通气、感染与炎症等,致使肺组织异常修复,产生持续性的肺部炎症,破坏了肺泡化和肺微血管发育,进而导致BPD 的发生[6-7]。最近有研究表明,NOD 样受体热蛋白结构域相关蛋白 3(nucleotide-binding oligomerization domain,leucine-rich repeat and pyrin domain-containing 3,NLRP3)炎症小体在BPD 的发展中起着至关重要的作用[8-10]。细胞在感受到病原体或损伤相关信号后,NLRP3 炎性小体被激活,通过凋亡相关斑点样蛋白招募并与Caspase-1 组装,导致Caspase-1 和IL-1β 的活化[11],这些信号事件最终通过促进机体产生持续性炎症反应,介导BPD 的发生发展。
细胞焦亡是一种程序性的炎性细胞死亡,由Caspase-1、Caspase-4 和Caspase-5(鼠Caspase-1 和Caspase-11)介导[12-13],是机体对感染和细胞损伤的免疫防御应答。消皮素D(gasdermin D,GSDMD)是细胞焦亡过程中的关键执行分子,经Caspase-1p20 切割成具有成孔活性的N-GSDMD[14],介导细胞焦亡的发生,然而目前NLRP3 介导的细胞焦亡在BPD 中的作用机制尚不明确。巨噬细胞是肺组织中固有免疫系统的重要成分,在高氧诱导的BPD中会向M1 炎性表型极化[15-16]。在高氧刺激下,炎性巨噬细胞是否通过细胞焦亡加剧炎症因子的大量释放,从而促进BPD 的发生,另外是否可以通过细胞焦亡调控BPD,目前尚不清楚。
本研究拟通过构建慢性高氧诱导的新生小鼠BPD 模型,旨在探讨NLRP3/Caspase-1/GSDMD介导的细胞焦亡是否在高氧诱导的BPD 中被活化,以及是否在巨噬细胞中被激活,同时观察细胞焦亡引发的炎症反应可能对肺泡上皮细胞造成的损伤。
材 料 和 方 法
试剂及仪器钙石灰(上海纳辉干燥试剂厂),制氧机(青岛海尔智家股份有限公司),亚克力密封箱(上海赣修像塑制品有限公司),测氧仪(杭州佳长电子科技有限公司),β-tubulin 抗体(美国Abcam公司),NLRP3 和Caspase-1 抗体(瑞士AdipoGen 公司),GSDMD 和IL-1β 抗体(美国Cell Signaling Technology 公司),CD31、F4/80 和AQP5 抗体(武汉赛维尔生物科技公司),TRIzol(美国Invitrogen 公司),反转录和qPCR 试剂盒(日本TaKaRa 公司),RIPA 裂解液、蛋白酶磷酸酶抑制剂和小鼠IL-1β ELISA 检测试剂盒(美国Thermo 公司),小鼠CD45(1∶200)和F4/80(1∶200)流式抗体以及阴阳磁珠(美国BD 公司),实时荧光定量PCR 仪(瑞士Roche 公司),电泳仪和转模槽以及化学发光成像仪(美国Bio-Rad 公司),流式细胞分析仪(美国BD 公司),光学显微镜和共聚焦显微镜(德国Leica 公司)。
高氧诱导的小鼠BPD 模型建立和分组实验动物为足月出生的新生小鼠,由C57BL/6J 孕鼠(孕16~18 天)自然分娩而来,孕鼠购自浙江维通利华实验动物技术有限公司。饲养于复旦大学附属浦东医院SPF 级动物房,饲养条件为室温22 ℃,12 h明暗循环,正常饮食饮水。本研究遵循实验动物饲养相关法律,得到复旦大学附属儿科医院动物研究委员会的批准(批准号:2020-64)并接受其监管。将40 只出生后24 h 内(postnatal days 0,PN0)的足月新生小鼠随机分为空气(room air,RA)组和高氧(hyperoxia,HO)组,每组20 只。高氧组小鼠连同母鼠饲养在密封的透明亚克力密封箱内,为保证密封箱内的高氧环境,进气口连通制氧机,用测氧仪监测氧气浓度至85%,箱内铺满钙石灰吸附CO2,保证箱内CO2浓度不超过0.5%。空气组小鼠及其母鼠放置于同一饲养室内空气环境中(21% O2)喂养(图1)。为避免母鼠因长期高氧刺激导致氧气中毒和喂养能力的下降,每隔24 h 高氧组与空气组交换哺乳母鼠。在出生后7 天(PN7)、14 天(PN14)的时间点处死小鼠,收集小鼠肺组织、血清以及支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)。

图1 慢性高氧诱导的BPD 小鼠模型设计图Fig 1 Schematic representation of chronic hyperoxia-induced BPD mouse model
实时荧光定量PCR 检测小鼠肺组织中促炎细胞因子的表达用剪刀剪取适量的肺组织装入匀浆管中,每50~100 mg 组织加1 mL TRIzol,用组织研磨仪搅匀组织样品。匀浆后将液体移到离心管中,静置5 min。接着按照TRIzol 试剂说明书抽提组织中的总RNA,根据操作说明书将RNA 逆转录成cDNA。依据目的基因序列信息设计并合成qPCR 引物序列(表1)。

表1 qPCR 引物序列Tab 1 qPCR primer sequence
以逆转录的cDNA 为模板,用内参基因和目的基因引物扩增管家基因和待测的目的基因片段,将样品置于实时荧光定量PCR 仪中,SYBR Green 法检测基因的表达,分析数据时采用2-ΔΔCT法(以βactin 为内参)。
蛋白免疫印迹(Western blot)在PN7、PN14的时间点处死小鼠,取肺,用PBS 冲洗后研磨彻底,4 ℃下12 000×g离心25 min,提取蛋白裂解液上清。使用BCA 试剂盒检测蛋白浓度。将等量蛋白质通过SDS-PAGE 分离,再转至PVDF 膜上。用5%的BSA 室温下封闭1 h,与一抗4 ℃ 孵育过夜:β-tubulin(1∶3 000),NLRP3(1∶1 000),GSDMD(1∶1 000),Caspase-1(1∶1 000),IL-1β(1∶1 000)。第二天用TBST 洗膜,与二抗室温孵育1 h,再次洗膜后用化学发光仪成像,用ImageJ1.8.0 软件进行灰度分析。
酶联免疫吸附试验(ELISA)颌下静脉采血,取PN14 小鼠的全血,室温下12 000×g离心25 min,得到血清。肺组织匀浆的上样量是100 μL/500 mg,血清稀释2 倍后上样,体积为100 μL。根据ELISA 试剂说明书进行操作即可。
HE 染色和Masson 染色取小鼠左肺组织放于4%多聚甲醛中,进行梯度酒精脱水。蜡块放置于石蜡切片机上切片,切片厚度5 μm,切片方向为横切气管方向。采用HE 染色和Masson 染色方法,用光学显微镜分析。辐射状肺泡计数(radial alveolar counts,RAC)是评估肺泡数目与结构简单化的指标。RAC 指的是从呼吸性细支气管中心到最近的纤维隔或胸膜垂直线上的肺胞数[17]。胶原沉积率是评估肺纤维化程度指标,利用ImageJ 1.8.0 软件统计数据。
免疫组化(immunohistochemistry,IHC)石蜡切片脱蜡至水,用柠檬酸抗原修复缓冲液(pH=6.0)进行修复,切片放入3%双氧水溶液,阻断内源性过氧化物酶,3% BSA 封闭,一抗4 ℃ 孵育过夜,HRP 标记二抗孵育50 min,DAB 显色,苏木精复染细胞核,脱水封片,白光显微镜检。肺微血管密度(mean microvessel density,MVD)是评估肺组织微血管发育程度的指标,应用Image-Pro Plus 6.0 软件进行图像分析。AQP5免疫组化相对定量用ImageJ 1.8.0软件分析。
组织样本免疫荧光(immunofluorescence,IF)石蜡切片脱蜡至水,EDTA 抗原修复缓冲液(pH=8.0)修复,画圈血清封闭,一抗4 ℃ 孵育过夜,二抗室温避光孵育50 min,DAPI 复染细胞核,加入自发荧光淬灭剂5 min,抗荧光淬灭封片剂封片,荧光显微镜下拍照,ImageJ 1.8.0 软件进行定量分析。
BALF 流式染色BALF 于4 ℃下400×g离心10 min,去上清,FACS缓冲液重悬;4 ℃下400×g离心5 min,去上清,流式抗体孵育30 min。同时制备空白管和各个抗体的单染管,孵育后加入FACS 缓冲液洗掉未结合的抗体;4 ℃下400×g离心5 min,去上清,FACS 缓冲液重悬,移入流式管,上机检测,FlowJo 10.8软件分析。
统计分析GraphPad Prism 9.0 软件用于可视化作图和数据统计学分析。实验数据以±s表示,P<0.05 为差异有统计学意义。计量资料采用正态检验和方差齐性检验。两组间的差异采用双尾t检验,多组间的比较采用方差分析(ANOVA)。多组间的两两比较采用Tukey 检验。
结 果
慢性高氧诱导的BPD 小鼠模型成功构建HE染色结果显示,7 天高氧组小鼠的肺组织有轻微病理改变,RAC 轻微减少,肺泡体积略增大。随着高氧的暴露时间延长,与对照组相比,14 天高氧组的肺组织形态逐渐发生改变,RAC 显著减少,肺泡体积明显变大,肺泡大小不均,提示肺泡化明显受损(图2A、2D)。CD31 免疫组化结果显示,7 天高氧组肺部MVD 稍低于正常空气对照组,而随着高氧的刺激时间加长,14 天高氧组的MVD 显著低于同期的空气对照组(图2B、2E),说明存在严重的肺微血管发育不良。Masson 染色结果表明,相比于对照组,7 天高氧组小鼠的肺组织未见明显病理改变,而14 天高氧组小鼠的肺纤维化程度显著增加,肺间质出现较多的胶原纤维增生,胶原沉积率明显提高,说明肺组织的纤维化主要发生在BPD 晚期(图2C、2F)。以上结果都说明高氧诱导的BPD 小鼠肺泡化阻滞,肺微血管生成障碍,肺纤维化程度显著增加,符合BPD 的病理特点,即BPD 小鼠模型成功建立。
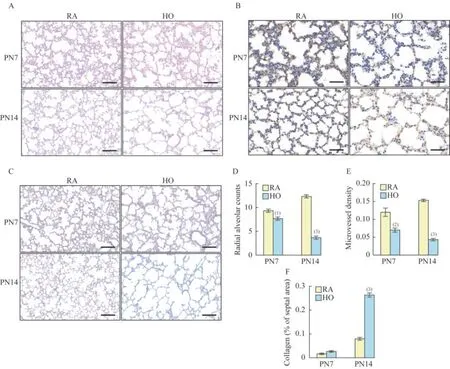
图2 慢性高氧诱导的BPD 小鼠模型建立Fig 2 Establishment of BPD mouse model induced by chronic hyperoxia
BPD 小鼠肺组织中促炎细胞因子表达上调为了研究慢性高氧诱导的BPD 小鼠肺组织中促炎细胞因子转录水平上的动态变化,我们通过qPCR检测小鼠肺组织的炎症因子Il6、Il1b、Mmp12和趋化因子Ccl2、Ccl5、Il8的表达。这些促炎细胞因子均与BPD 的发生密切相关[18-21]。我们发现与同期对照组相比,Il6、Mmp12和Ccl2mRNA 在7 天和14 天的高氧组小鼠中都显著升高,提示这些促炎因子的表达是稳定升高的,一直持续到BPD 小鼠模型结束;而Il1b、Ccl5和Il8mRNA 在7 天高氧组小鼠中显著上调,但在14 天高氧组小鼠中却已经无明显差异,提示这些促炎因子的转录水平高峰期可能发生在BPD 小鼠模型的早中期(图3A~3F)。以上结果表明慢性高氧诱导的小鼠BPD 中,促炎细胞因子的mRNA 表达水平明显受到严格的时空调控,有助于我们选择合适的时间点进行精准干预。
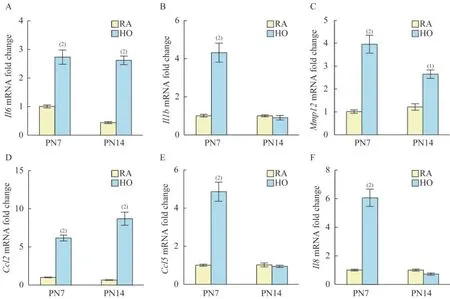
图3 各组小鼠肺组织中促炎细胞因子和趋化因子的mRNA 表达Fig 3 The mRNA expressions of pro-inflammatory cytokines and chemokines in lung tissue of mice from each group
BPD 小鼠的BALF 中巨噬细胞比例升高收集14 天各组小鼠的BALF,进行流式抗体染色。CD45标记白细胞,F4/80 标记小鼠巨噬细胞。流式分析结果显示,高氧组CD45+F4/80+双阳性的小鼠巨噬细胞比例显著升高(图4A、4B),说明高氧组小鼠肺组织中巨噬细胞浸润增多,可能参与了肺部的炎症反应。
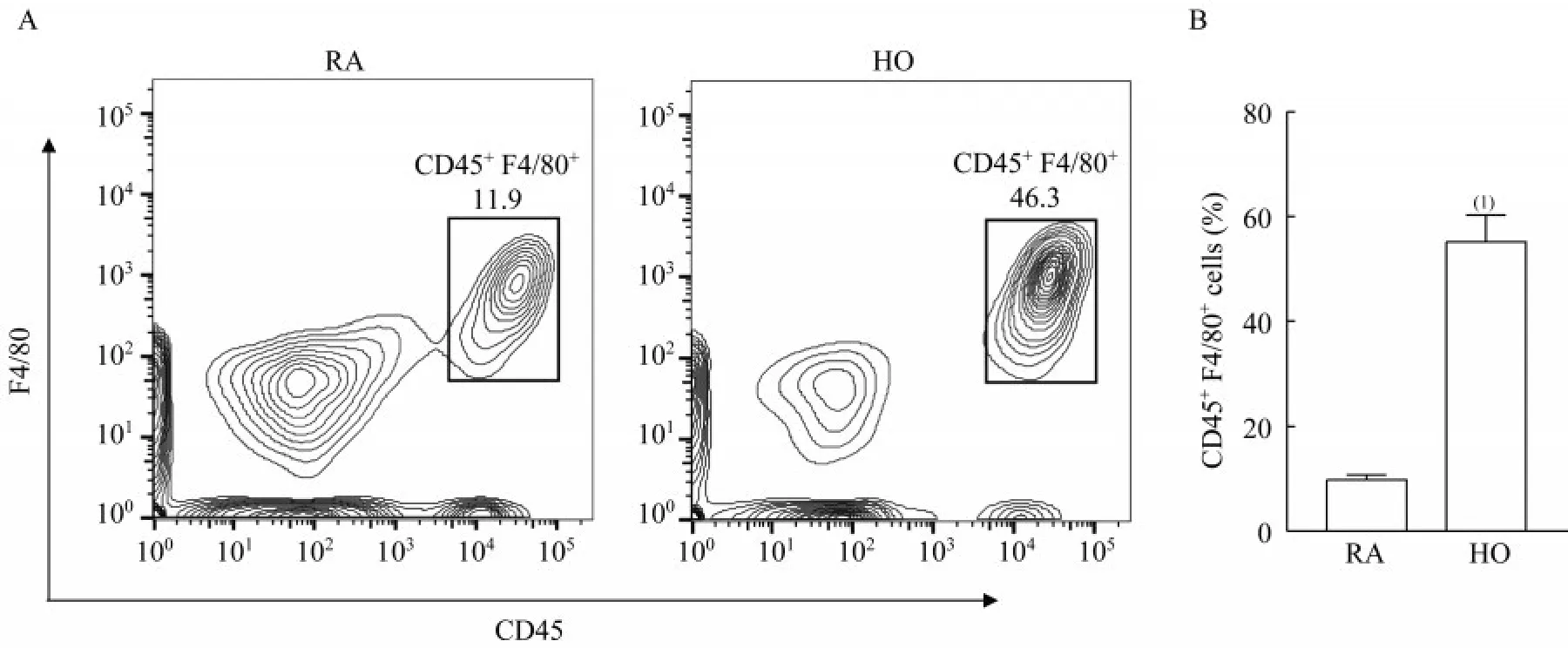
图4 各组小鼠的BALF 中巨噬细胞比例Fig 4 Proportion of macrophages in BALF of mice from each group
NLRP3/Caspase-1/GSDMD 介导的细胞焦亡在BPD 中被激活为进一步研究NLRP3/Caspase-1/GSDMD 介导的细胞焦亡是否在BPD 中被活化,我们利用Western blot 分析各组小鼠肺组织中NLRP3、核心成分Caspase-1、焦亡执行蛋白GSDMD 以及炎症因子IL-1β 的表达水平。结果显示,与14 天空气对照组相比,14 天高氧组中NLRP3炎症小体明显活化,Caspase-1 p20 的表达显著增高,N-GSDMD 的剪切大量增加,成熟的IL-1β 分泌显著增多,而对照组小鼠几乎检测不到(图5A、5B)。提示BPD 小鼠肺组织中NLRP3/Caspase-1/GSDMD 介导的细胞焦亡被激活,加重了肺部炎症反应,可能参与了BPD 的发病。
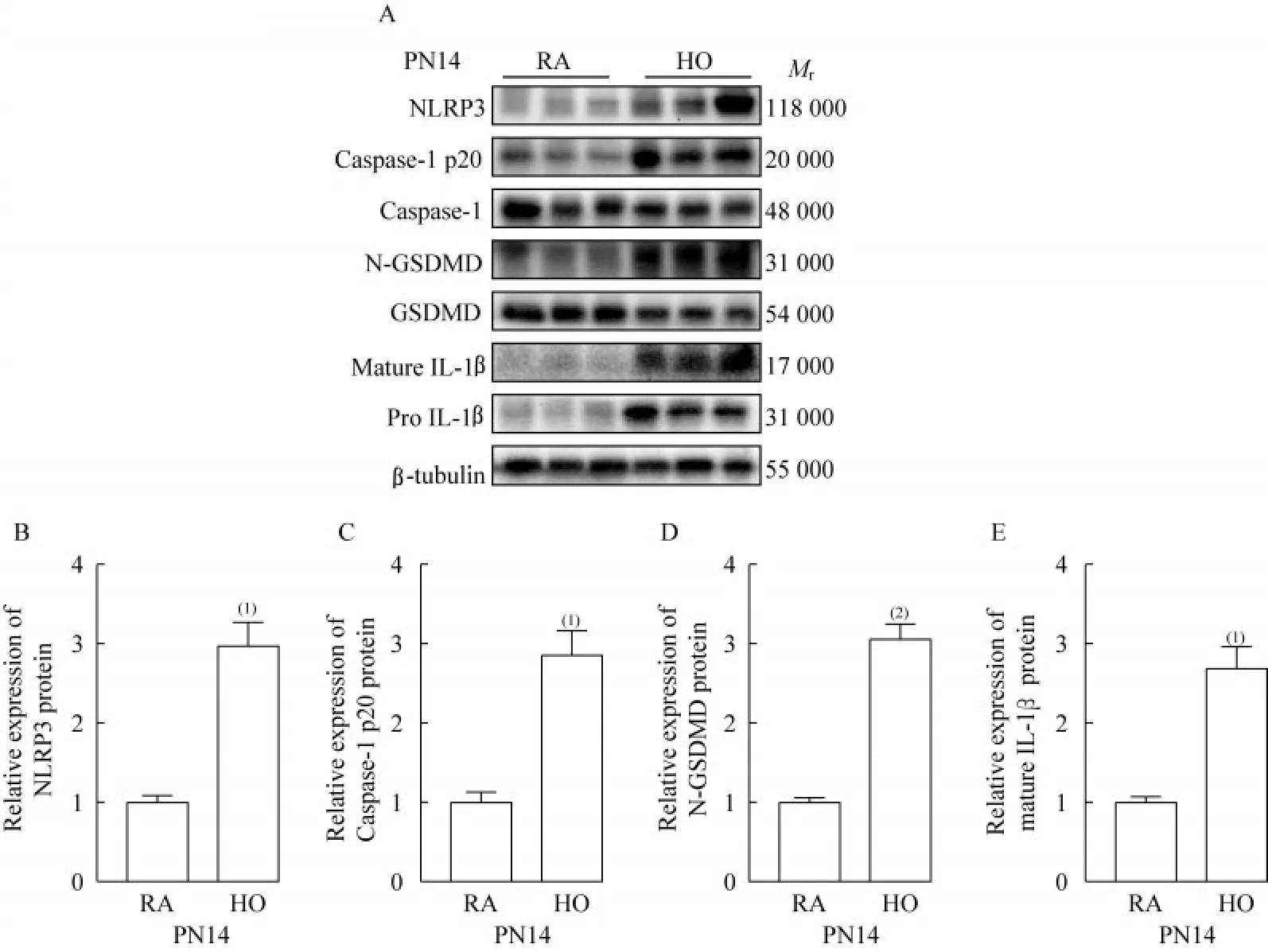
图5 各组小鼠肺组织中细胞焦亡相关蛋白的表达水平Fig 5 Expression levels of pyroptosis-associated proteins in lung tissues of mice from each group
NLRP3/Caspase-1/GSDMD 介导的焦亡在巨噬细胞中显著增强为探究巨噬细胞焦亡是否在BPD 中被激活,我们使用了免疫荧光共定位技术对14 天各组小鼠肺组织进行双染色,观察F4/80 分别与细胞焦亡相关蛋白NLRP3、Caspase-1 以及IL-1β的共定位情况。F4/80 是小鼠巨噬细胞经典的生物标志物。结果显示,与空气对照组相比,高氧组F4/80 阳性的巨噬细胞数量明显增加,即BPD 小鼠肺组织中巨噬细胞浸润增多,而且高氧组NLRP3、Caspase-1、IL-1β 与F4/80 都存在显著的共定位现象,双阳性细胞数量明显增多,荧光共定位信号大大增强(图6A~6F)。这些情况说明在BPD 小鼠肺组织中巨噬细胞焦亡被激活,且焦亡现象十分明显。

图6 免疫荧光检测各组小鼠肺组织中NLRP3/Caspase-1/IL-1β 与F4/80 的共定位Fig 6 IF co-localization of NLRP3/Caspase-1/IL-1β with F4/80 in lung tissue of mice from each group
BPD 小鼠肺组织匀浆和血清中IL-1β 的水平显著升高为进一步评估小鼠机体的炎症状态,我们使用ELISA 检测了各组小鼠肺组织匀浆和血清中IL-1β 的表达。结果显示,相较于14 天空气对照组,14 天高氧组小鼠肺组织匀浆和血清的IL-1β 均有显著增加(图7A,7B),表明持续高氧刺激下,BPD 小鼠肺组织的炎症亢进,细胞焦亡过度激活,可能致使IL-1β 等促炎因子大量产生并释放到外周血中,机体处于一个炎症过强的状态。
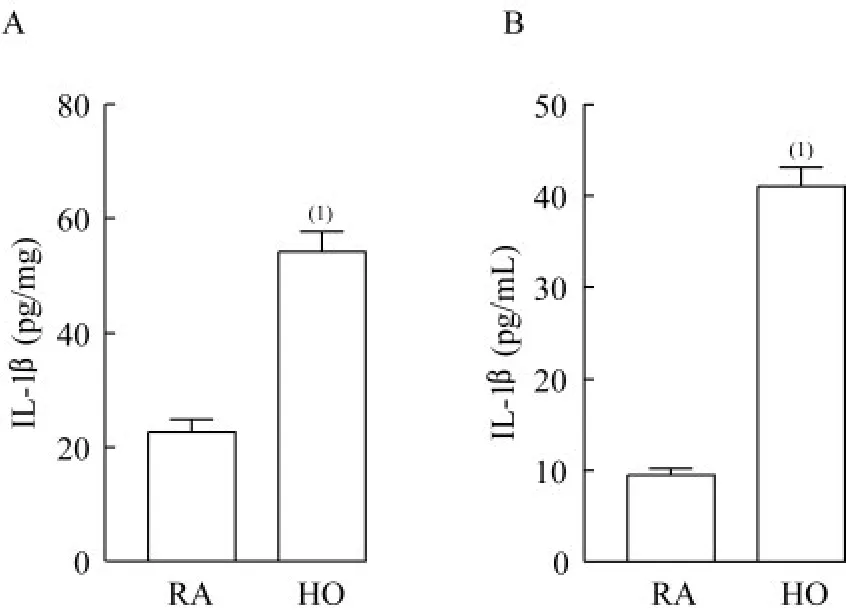
图7 ELISA 检测各组小鼠肺组织匀浆和血清中IL-1β 的水平Fig 7 Concentrations of IL-1β in lung homogenate and serum of mice from each group
BPD 小鼠的肺泡上皮细胞严重受损肺泡Ⅰ型上皮细胞(alveolar epithelial type Ⅰ,AT Ⅰ)是维持正常的肺泡气体交换功能所必需的,是肺泡上皮的重要成分,也是数量最多的肺泡上皮细胞。AQP5 是ATⅠ的生物标志物。qRT-PCR 结果显示,空气组小鼠肺组织的Aqp5mRNA 水平随着生长的日龄逐步升高,高氧组在7 天时还未见明显变化,在高氧的不断刺激下,至14 天时,Aqp5mRNA水平下降,显著低于同期的空气组(图8B)。免疫组化结果显示,14 天空气组小鼠肺组织中AQP5 存在明显的高表达,而14 天高氧组中AQP5 表达显著下降(图8A、8C)。这说明高氧环境中所产生的促炎性因子很可能会损伤ATⅠ,AT Ⅰ数量减少,肺泡上皮功能下降,阻碍了肺泡的生长发育,促进了BPD 的发病。

图8 各组小鼠肺组织中Aqp5 mRNA 和AQP5 蛋白的表达水平Fig 8 Expressions of Aqp5 mRNA and AQP5 protein in lung tissue of mice from each group
讨 论
人和小鼠的肺发育进程相似,人的肺发育时期分为胚胎期、胎儿期(假腺期、小管期、终末囊泡期)和肺泡期,而小鼠分为假腺期、小管期、囊泡期、肺泡期和成熟期[22-23]。早产儿是指胎龄达28 周但不足37 周的婴儿,此时婴儿的肺发育处于囊泡期[24],而小鼠肺发育的囊泡期发生在胚胎第17.5 天至生后第5 天[25],与新生小鼠(孕21 天至出生后4 天)的肺发育处于同一时期,故足月新生小鼠是天然的人类早产动物模型,可作为BPD 实验模型的研究对象。人类的终末囊泡期对应于人肺发育的孕24~36周[26],大部分早产儿的肺出生时刚好处于终末囊泡期,肺泡化还未成熟,对外部刺激特别敏感,尚未健全的肺受到高氧或其他不利因素损伤,就会导致BPD 的发生发展[27]。
本研究将新生小鼠暴露在85%氧气中,在出生后7 天、14 天两个时间点采集数据和观察各项指标。PN7 高氧组可观察到轻度的肺泡化阻滞,RAC 轻微减少,肺泡体积略大,而PN14 高氧组的肺泡化严重受损,肺泡体积明显增大,大小不均,部分肺泡融合,说明肺泡化进程严重受阻,这与文献报道一致[28]。CD31 免疫组化结果提示,高氧组肺组织的微血管生成也在PN7 开始减少,直至PN14 出现严重的肺微血管发育不良,这也符合BPD 的第二大重要病理特点[29]。HE 染色和CD31 组化结果说明BPD 的肺泡化阻滞和肺微血管生成障碍在早期即可出现明显病变,也是BPD 最主要的两个病变特征,符合“新型”BPD 的特点[30]。而Masson 染色结果提示高氧早期肺纤维化并未见明显改变,而是到了高氧后期才出现显著的胶原纤维增生,这些结果说明BPD 肺组织的远期纤维化会加重不良预后,也符合“经典”BPD 的特点[31]。本研究结果表明出生后新生小鼠暴露在85%的氧气中持续14 天,导致了肺损伤。从病理形态学上观察,该肺损伤的表型基本与人类“经典”BPD 的异常组织病变一致。这表明本实验的慢性高氧诱导的BPD 小鼠模型是成功的。
近期研究报告NLRP3基因的敲除或抑制NLRP3 炎性小体活化可以保护高氧诱导的小鼠BPD[8,32-33]。在临床研究中,早产儿支气管抽吸物中IL-1β 水平升高与BPD 发病率增加相关[34-35]。我们同样观察到BPD 小鼠肺组织中NLRP3 炎性小体组装增加,肺组织匀浆和血清中成熟IL-1β 的释放增多,而且其上游的Caspase-1 p20 裂解片段增加,NGSDMD 的剪切活化,说明NLRP3/Caspase-1/GSDMD 介导的细胞焦亡在高氧诱导的BPD 小鼠肺组织中明显被过度激活,可能加重了肺部炎症反应。而且在PN7 即可发现Il1b、Il6和Mmp12等促炎因子的mRNA 表达升高,表明炎症在BPD 早期即参与疾病的发生发展,可能是介导远期肺纤维化的重要病因。既往研究表明,BPD 患者的气管抽吸物中中性粒细胞和巨噬细胞增多[35-38]。我们的流式分析结果也显示BPD 小鼠BALF 中巨噬细胞比例显著升高,而且在PN7 即可见肺组织中单核-巨噬细胞趋化因子Ccl2和Ccl5的mRNA 表达升高,中性粒细胞趋化剂Il8的mRNA 表达上调,这进一步说明巨噬细胞和中性粒细胞也许在BPD 早期即可浸润到受损的肺组织中,参与肺部炎症反应。
巨噬细胞是炎症性疾病进展过程中的核心介质,也是肺部先天免疫的重要细胞,参与炎症的起始和消除。最近有研究结果显示[18],高氧可激活肺中未成熟的巨噬细胞,促进炎症因子的释放,通过IL-6/STAT3 轴损伤肺泡上皮细胞的再生位点,抑制肺生长发育。目前高氧如何导致巨噬细胞大量释放炎症因子的方式尚不明确。基于NLRP3 介导的细胞焦亡在BPD 肺组织中被激活这一现象,因此我们进一步通过F4/80 与NLRP3、Caspase-1 和IL-1β 的3 个免疫荧光共定位确定了BPD 小鼠肺组织中大量浸润的巨噬细胞发生焦亡。这可能加速了炎症因子的过度释放,从而促进BPD 的发生发展。此外,我们观察到高氧严重损伤了BPD 小鼠的肺泡上皮细胞,导致肺泡上皮功能障碍。因此,我们推测高氧诱导的巨噬细胞焦亡所造成的肺部“高炎”环境也许在一定程度上损伤了肺泡上皮细胞,但仍需要更多的体内和体外共培养实验进一步证实。
虽然我们证明了高氧诱导BPD 小鼠肺组织中巨噬细胞焦亡被激活,但是仍然不能排除其他非免疫细胞如上皮细胞或者内皮细胞也发生焦亡的可能性,即各种细胞的焦亡现象都可能参与了BPD 的肺部炎症反应。细胞焦亡主要是一种先天免疫反应,巨噬细胞是固有免疫系统的核心成分,而且也有研究表明肺组织的巨噬细胞在BPD 中对免疫调节功能至关重要[39-41],故我们可能更倾向于考虑固有免疫细胞(即巨噬细胞焦亡)起主导作用,而非肺部的实质细胞。中性粒细胞也是固有免疫系统的重要成分,同样参与BPD 肺部的炎症反应[42],但大多数有关GSDMD 诱导细胞焦亡的研究集中在巨噬细胞上,尽管有研究表明GSDMD 对中性粒细胞发挥功能也有一定作用,但是与巨噬细胞相反,中性粒细胞内通过NLRP3 激活的GSDMD 并不容易导致焦亡[43],而是通过靶向细胞核促进中性粒细胞胞外杀菌网络(neutrophil extracellular traps,NET)的形成[44]。最近也有研究报道证实,中性粒细胞的确可以通过NET 促进BPD 的肺损伤[45-46]。
综上所述,我们确定了NLRP3/Caspase-1/GSDMD 介导的巨噬细胞焦亡很可能参与了BPD的发病,是一个重要的治疗靶点。未来将进一步对NLRP3/Caspase-1/GSDMD 介导的巨噬细胞焦亡通路进行药物干预或者靶向抑制,以期为BPD 的肺损伤提供新的治疗策略。
作者贡献声明涂子坤 实验设计,数据分析,论文撰写。高雅静 实验设计,论文修订。韩晓 研究指导,数据分析。郭海艳 数据分析。周玉峰研究设计和监督指导,论文修订。
利益冲突声明所有作者均声明不存在利益冲突。
猜你喜欢
中国医学科学院学报(2023年1期)2023-10-15
世界科学技术-中医药现代化(2022年3期)2022-08-22
医学综述(2022年7期)2022-04-19
食品工业(2021年12期)2021-12-31
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年4期)2021-07-31
昆明医科大学学报(2020年12期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际呼吸杂志(2019年8期)2019-04-29
国际呼吸杂志(2019年8期)2019-04-29
