
制氢光催化剂钴配合物的理论研究
2023-12-13 01:15石海霞苗体方
淮北师范大学学报(自然科学版) 2023年4期
李 双,石海霞,苗体方
(淮北师范大学 化学与材料科学学院,安徽 淮北 235000)
0 引言
在当今世界,能源危机成为各个国家所面临最主要的问题,也是地球所面临的两大难题之一。发展可持续的清洁能源,如水电、风能和太阳能等可以缓解能源危机和大量使用化石能源造成的环境问题。太阳能取之不尽、用之不衰,科学家通过光催化分解水,将太阳能转换为高热值的H2。自Fujishima 和Honda首次报道关于TiO2光催化分解水产生氢气以来,具有高催化活性、良好的稳定性、对人体无毒性和低成本的金属钛化合物就已经成为光催化领域中研究和应用最广泛的金属材料之一。近年来,一系列其他光催化剂金属化合物被开发出来,其中,钴配合物由于成本低、性能稳定和应用前景广泛而成为众多科学家的研究对象。Eisenberg等[1]报道含有单分子钴肟配合物[Co(dmgH)2pyCl],用三联吡啶Pt(II)作为光敏剂,乙醇胺为牺牲剂,在pH 值为8.5 条件下,体系的制氢催化循环数(TON)达到1 000(vs,催化剂)。Thummel等[2]报道1例以[Ru(bpy)3]2+为光敏剂,钴配合物为催化剂,抗坏血酸为牺牲剂,该体系在pH值为4 的溶液中,经过3 h 的光照TON 达到333(vs,催化剂)。Mcnamara 等[3]合成一个二硫纶配合物[Co(bdt)2]-,[Ru(bpy)3]2+为光敏剂,抗坏血酸为牺牲剂,该体系经过3 h的光照,TON达到2 700(vs,催化剂)。Chakraborty等[4]合成一个钴配合物CoGGH,[Ru(bpy)3]2+为光敏剂,抗坏血酸为牺牲剂,体系的制氢活性能维持48 h,TON达到2 200(vs,催化剂)。可见,光催化剂钴配合物在解决能源危机方面具有潜在的应用前景。
密度泛函理论(DFT)方法是基于量子力学方法开发的,可以作为研究电子结构和催化性能之间的桥梁,对光催化剂钴配合物结构研究不仅有助于理解光催化剂结构-性能关系[5],还可以预测催化剂性能。本文主要是利用密度泛函理论研究不同光催化剂钴配合物的电子结构,从理论上预测钴配合物光催化分解水的性质,其结构式如图1所示。

图1 钴配合物1~3的结构示意图和部分原子编号(不显示部分氢原子)
1 计算方法
由于钴配合物受水溶液影响较大,在B3LYP/6-31G(d)水平上,在水溶液中采用连续极性导体模型(CPCM)对配合物进行优化[6]。以优化的几何结构为基础进行频率计算,计算结果表明,所有频率都是正值,表明优化得到的3个配合物几何结构都是稳定结构。在此结构基础上采用TDDFT(含时密度泛函理论)方法,计算260个单重激发态,用GaussSum 2.0软件模拟出电子吸收光谱图。对配合物1~3前线分子轨道进行计算,并模拟出配合物最高占据轨道(HOMO)和最低未占据轨道(LUMO)的分子轨道图。以在水溶液中优化的结构为基础,在真空中进行计算,获得配合物在真空中的能量,采用基于密度的溶剂模型(SMD)[7]获得配合物在水溶液中的能量,根据文献[8]计算出钴配合物还原电势。所有计算由Gaussian 16完成。
2 结果与讨论
2.1 计算方法准确性评估
配合物1~3都是以钴为中心原子的配合物,都可以作为光催化制氢的催化剂,由于钴配合物中钴离子易受水溶液影响,因此,钴配合物的优化考虑水溶液是必要的。由于配合物2有实验数据,在水溶液中优化配合物2得到的部分数据列于表1,从表1中的数据可以看出,在水溶液中优化得到的配合物结构和实验结果吻合较好,用此方法优化得到的钴配合物结构是可信的。
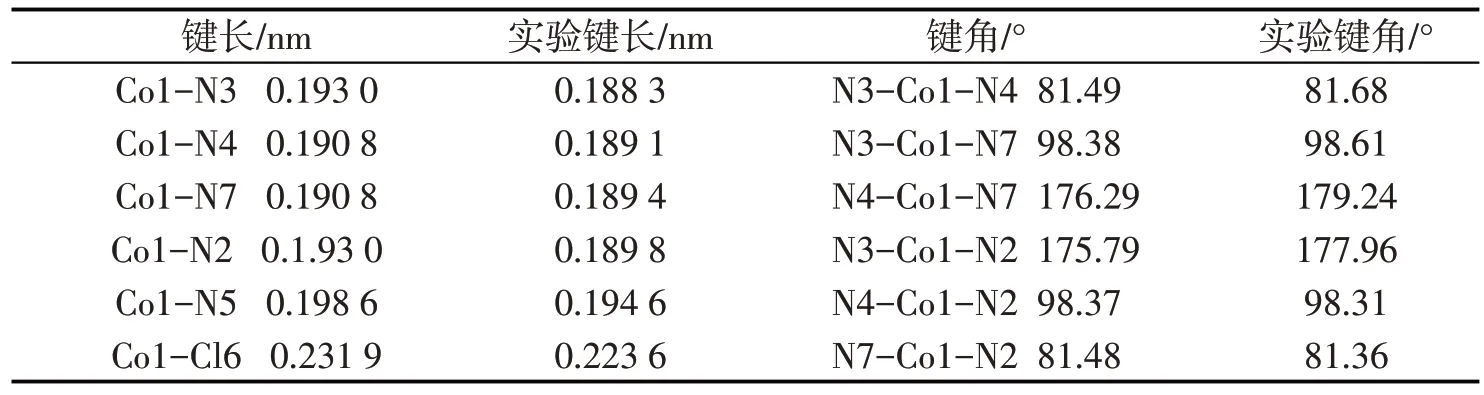
表1 配合物2部分结构参数和相应的实验值
以在水溶液中优化配合物2的结构为基础,在水溶液中计算配合物2的电子吸收光谱[9-10],并进行模拟,结果见图2。从图2 可以看出,配合物2有2个强的吸收峰,分别位于253.24 nm和189.34 nm,实验结果大约位于256 nm和204 nm,无论是2个强吸收峰的位置还是强度都和实验结果吻合较好[11],进一步说明此方法适合钴配合物的计算,这为研究钴配合物光催化制氢性质奠定坚实的理论基础。

图2 计算得到的配合物2的电子吸收光谱
2.2 还原电势分析
在光的激发下,配合物和光敏剂之间发生分子间电子转移,从而导致配合物在水溶液中有氢气释放,这是因为光敏剂被激发,电子从光敏剂向光催化剂配合物转移,配合物获得电子后,其周围的氢离子很容易从配合物上获得电子而释放氢气,所以配合物和光敏剂之间的电子转移对氢气的释放起着重要作用。电子转移的实质就是光敏剂具有高的氧化电势,配合物具有低的还原电势,配合物的还原电势越低,配合物越易得到电子,配合物的制氢活性越好。为验证这一想法,计算配合物1~3 的还原电势,分别为-0.342 0 V、-0.153 2 V和-0.276 3 V,可以看出配合物1的还原电势最低,配合物1容易获得电子,配合物1的制氢活性好于配合物2,与实验结果相符合[11]。
2.3 前线轨道分析
以优化的几何结构为基础,对配合物1~3 的前线分子轨道进行计算,得到配合物的最高占据轨道(HOMO)和最低占据轨道(LUMO),并模拟出配合物的HOMO和LUMO图,结果见图3。
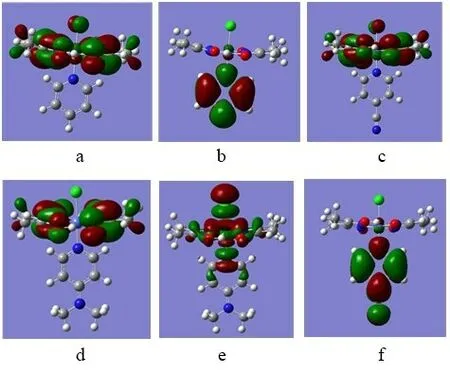
图3 配合物1~3的HOMO和LUMO轨道
从图3可以看出,配合物HOMO 上的电子云主要分布在金属钴周围,LUMO 上的电子云主要分布在吡啶环和氯原子上。在光照射下,配合物上的电子主要从金属钴周围的原子向吡啶环和氯原子转移,或许这样一种电子转移方式都有利于氢气释放。从图3还可以看出,在光照射下,配合物1和2电子转移路径是不同的,配合物1上的电子主要从金属钴周围的原子向吡啶环转移,在吡啶环上聚集较多的电子,其周围氢离子容易获得电子释放氢气,而配合物2上的电子主要从金属钴周围的原子向氯原子转移,氯原子上聚集大量电子,由于氯原子电负性较强,氯原子上的电子很难和周围氢离子结合释放氢气,这样会导致配合物2制氢活性低于配合物1,与实验结果吻合较好[11]。
尽管3个配合物中心金属相同,由于配体的差异,导致电子云分布有较大差异。配体和官能团的改变会影响电子转移方向,进而影响配合物制氢活性[12-15],对于设计高性能催化剂具有一定的理论参考价值。
3 结论
采用DFT方法,对3个钴配合物进行理论计算,准确评估钴配合物的理论计算方法,获得3个配合物的稳定结构,并计算这3个配合物的还原电势和前线分子轨道,探索配合物结构和制氢活性的关系,从理论上预测配合物1的光催化制氢性能好于配合物2和3,希望这些研究结果为设计、合成高性能光催化剂及探索光催化反应机理提供理论参考。
猜你喜欢
化工学报(2021年1期)2021-01-30
防爆电机(2020年4期)2020-12-14
河北理科教学研究(2020年1期)2020-07-24
山东化工(2019年2期)2019-02-16
福建农林大学学报(自然科学版)(2018年5期)2018-10-11
浙江大学学报(工学版)(2016年9期)2016-06-05
中学生理科应试(2016年2期)2016-05-30
广东石油化工学院学报(2016年3期)2016-05-17
应用化工(2014年8期)2014-08-08
中国药理学通报(2014年2期)2014-05-09
