
KIRC组学数据分类的自注意亚型识别神经网络
2023-12-11 07:11陈锡程伍亚舟
计算机工程与应用 2023年23期
李 阳,陈锡程,伍亚舟
陆军军医大学 军事预防医学系 军队卫生统计学教研室,重庆 400038
癌症是一种高度异质性的疾病,其不同类型的癌细胞具有不同的生物学特征和表型[1]。这意味着即使是同一种癌症,在不同患者中也可能会表现出不同的亚型[2]。这种异质性对于癌症的治疗和预后都有很大的影响[3]。以肾细胞癌为例,肾透明细胞癌(kidney renal clear cell carcinoma,KIRC)是肾细胞癌中最常见的病理类型[4]。虽然手术或消融等治疗方法可以治疗KIRC,但仍有约30%的患者可能发生转移,这会大大影响治疗效果和预后[5]。更深入地理解癌症亚型可以帮助医生制定更加个性化的治疗方案,提高治疗效果和患者的生存率[6]。因此,对于癌症亚型的认识对于癌症患者的治疗选择和预后具有重要意义。
近年来,随着高通量测序技术的发展,产生了大量的癌症基因组数据。这些数据可通过一些以癌症基因组图谱(the cancer genome atlas,TCGA)为代表的公开项目中获取[7]。随着精准医疗概念的兴起,对患者进行个性化的诊断和治疗,利用这些数据确定KIRC 的亚组分型将会对其早发现早治疗发挥积极的作用[8]。组学数据分析有利于预测癌症亚型,并提高对癌症进展的理解。因此,迫切需要对癌症亚型识别中的组学数据进行有效分析。然而,既往研究中利用基因组学数据对亚型确定的研究较多集中在乳腺癌[9]、肺癌[10]和肝癌[11]等癌症,对于肾细胞癌的亚型确定研究较少。
随着人工智能技术的不断发展,机器学习和深度学习已经广泛应用于生物医学领域。自注意力机制是一种用于处理序列数据的机制,其最早在Transformer 模型中被提出[12]。通过自注意力机制,模型能够更好地获取序列数据之间的关系,从而提高模型的性能[13]。自注意力机制已经广泛应用于自然语言处理和计算机视觉等领域。基于注意力的自编码器更适合学习高级功能[14]。尽管注意力机制主要在医学图像中有所应用[15],但在癌症测序数据中仍有待进一步研究。
因此,本文提出了一种新的深度学习模型自注意力亚型识别神经网络(self-attention subtype recognition neural network,SSRNN),并在KIRC组学数据分类中取得了优异性能。主要贡献和创新点可总结如下:(1)本文提出了一种新颖的深度学习方法SSRNN,它将编码器、自注意力机制、解码器和分类器结合到一个统一的框架中。这种方法可以有效提升模型的表现。(2)在基于深度学习的癌症亚型识别任务中首次引入了自注意力机制。通过这种方法,模型能够自动地学习样本之间的相似性,从而更好地表示数据特征。(3)在KIRC 的转录组学数据集上进行了实验,并采用非负矩阵分解的方法确定了KIRC的亚型分组。
1 数据收集和分析
全文数据和方法的技术路线如图1所示。

图1 全文技术路线图Fig.1 Full-text technology roadmap
1.1 数据收集
TCGA 是由美国国家癌症研究所和美国国家人类基因组研究所共同构建的项目,其中收入了各种癌症的临床数据、基因组数据等,是重要的癌研究的数据来源[7]。从TCGA的KIRC数据库(TGCA-KIRC database)中下载转录组学数据、临床数据和样本质量注释文件。
通过查看注释文件并排除低质量样本后,获得了594 例转录组学数据样本,其中523 例为KIRC 样本,71例为正常样本。从中提取了蛋白编码基因的Counts 数据(read Counts)和TPM 数据(transcripts per million)。其中Counts 数据是指在高通量测序每个基因在样本中被检测到的次数,TPM 数据是按每百万总读数为单位对每个基因在样本中的表达量进行归一化处理。
此外,还获取了537例临床数据。临床数据中提取了对应样本的年龄、性别、肿瘤分级、肿瘤分期、TNM分期、生存时间和生存状态的临床信息
依据样本ID 将转录组数据和临床数据取交集,最终获取514例癌症样本,这些样本将会被用于癌症亚型的确定和模型构建。
样本质量注释文件中将用于评估样本的质量,有助于筛选出高质量的样本进行分析。鉴于数据可能存在批次效应,从UCSC Xena 网站(https://xenabrowser.net/datapages/)下载了TCGA-KIRC的批次效应文件以去除批次效应,并提高数据的准确性。
1.2 生信分析
本文的生物信息学分析主要包括差异分析和富集分析。
为更深入地了解KIRC 的基因表达特征,对蛋白编码基因进行了差异分析,筛选出差异表达的基因。在差异分析前,对样本进行了过滤筛选,利用注释文件去除低质量的样本。同时,针对蛋白编码基因表达数据中存在的重复基因,选取较大的值作为该基因的表达值,以确保数据的准确性。其次,将数据分为KIRC 组和正常样本组,并结合批次效应文件,去除批次效应的影响。最后,利用R 语言中的DESeq2 包对蛋白编码基因数据进行差异分析,其中参数以差异倍数(lbFC)为1,校正p值(padj)为0.05 进行设置。通过差异分析获取了差异表达的基因,并根据差异分析结果绘制了火山图,以直观地展示基因的表达差异情况。
在获得了差异基因后,进一步对差异基因进行了富集分析,以更深入地了解这些基因在生物过程中的功能和作用。采用了GO(gene ontology)富集分析和KEGG(kyoto encyclopedia of genes and genomes)富集分析两种方法[16]。GO 富集分析可了解基因在分子功能、细胞组分和生物过程等方面的作用,而KEGG则是一个包含生物系统功能和化学信息的数据库,可帮助了解基因在哪些通路上具有富集[17-18]。在进行富集分析时,使用了R语言中的clusterProfiler包,以便有效地处理和分析数据。
1.3 聚类分析
将差异基因与临床的生存数据结合筛选出与KIRC患者生存相关的蛋白编码基因。共有514 例癌症样本纳入。利用R语言进行Cox单因素分析,设置p<0.001,由此筛选出与生存相关有意义的基因,最终得到生存相关的蛋白编码基因及其表达数据。
在获取与KIRC 生存相关的蛋白编码基因数据后,本文采用了一种数据降维的方法——非负矩阵分解(non-negative matrix factorization,NMF)以确定KIRC的亚型分类数量[19]。使用NMF算法执行聚类分析和降维的主要优势在于:(1)NMF算法是一种非负矩阵分解方法,可以将原始数据进行降维,同时得到一组非负的基向量和系数矩阵,这些基向量和系数矩阵可以用于描述原始数据的特征和结构。(2)相较于其他聚类技术,NMF 具有更大的性能优势,在癌症类别发现中提高了聚类结果[20]。(3)NMF 算法可以保留原始数据的非负性和稀疏性,能够更好地反映生物学系统的特征。(4)NMF可从基因表达数据中提取数据内部的特征,从而实现对样本进行分组的目的[21-22]。
具体而言,基于R语言中的NMF包对KIRC蛋白编码基因的非负矩阵进行分解。在参数设置方面,使用了上述获得的与生存相关的蛋白编码基因表达数据,采用默认的brunet 方法,聚类数k设置为2~10。NMF 包处理会输出Cophenetic 指数和一致性矩阵(consensus matrix)。Cophenetic 指数是一种常用的聚类质量评估指标,用于评价聚类结果的好坏[23]。Cophenetic 指数越接近于1,表示聚类效果越好,反之则聚类效果越差。一致性矩阵则用于评价非负矩阵分解结果的稳定性和可靠性,从中选择最优的分解结果[24]。
1.4 生存分析
根据NMF 包的处理结果,本文确定了最优的亚型分类数量,并将分类结果与生存数据结合,绘制生存曲线图以比较不同分型之间的生存情况是否存在差异。
2 模型建立和训练
本文提出了一种自注意力亚型识别神经网络(selfattention subtype recognition neural network,SSRNN),现将模型构建、训练和评价过程阐述如下。
2.1 特征选择
基因表达数据的维度问题是一个普遍存在的问题,即数据样本较少而数据特征过多。过多的数据特征容易包含更多的冗余信息,而这些冗余信息可能会导致模型的过拟合[25]。因此,需对数据特征进行筛选以提高模型的准确性和稳定性。
具体而言,对于KIRC的蛋白编码基因表达数据,需首先对其进行了Z-score归一化处理以消除数据特征之间的量纲差异。接着,对于归一化处理后的数据,使用方差过滤法对特征进行筛选,以去除方差为0或较小的特征,从而减少冗余信息的影响[26]。最后,本文采用了卡方过滤的方法,选取卡方值排序前100 个特征,并将其作为最终输入模型的特征[27]。这可进一步减少特征数量,去除冗余信息,并提高模型的预测准确性。下面简述100 个特征的设置原因。结合研究的样本数量和参考既往研究[28],当选择特征较少时可能引发效果较差,当选择特征过多时可能使效果提升不明显,因此最终选择100个特征,这有利于规避基因数据普遍存在梯度爆炸问题。
在上述处理过程中,均利用Python 进行数据的处理,分别使用了sklearn.preprocessing.MinMaxScaler 和sklearn.feature_selection 中的VarianceThreshold、chi2、SelectKBest。
2.2 模型建立
针对预处理后的KIRC 数据,本文构建了基于自注意力机制的KIRC亚型分类模型。构建的模型整体上分为两部分,即编码器部分和分类器部分,模型结构如图2所示。构建模型的过程中,需要考虑到癌症分型样本存在不均衡的问题。为了解决这个问题,采用了Pytorch中的WeightedRandomSampler 对样本进行加权处理。WeightedRandomSampler是一种能够对样本进行加权的随机采样器,它给每个样本分配不同的权重,以保证每个类别的样本均可得到训练以规避过拟合风险。

图2 SSRNN框架结构Fig.2 SSRNN framework structure
在编码器部分,采用了自编码器模块和自注意力模块两个模块来对数据进行特征提取和降维处理。自编码器模块是一种无监督学习的方法,它可以将输入数据压缩成低维表示,从而去除数据中的冗余信息。通过训练自编码器,可以得到一个压缩后的特征空间,从而实现对数据的降维处理。
为了更好地构建自注意力模块的数据矩阵,采用了数据降维的方法,将原始数据从100个维度降维到30个维度。这样可以减少数据的复杂性,并更好地利用数据信息。经过自编码器的处理后,每个样本都被表示为一个具有30个维度的向量。自注意力机制是一种能够计算相似度、考虑各元素之间关系的机制,可以更好地获得全局信息。通过计算每个元素与其他元素的相似度,自注意力机制可以计算出序列中各元素的权重矩阵,并利用该权重矩阵得到原数据的新表示。
在本研究构建的自注意力机制模块中,将每个样本降维后的30 个维度数据构建为一个3×10 的数据矩阵X。将该数据矩阵线性映射出三个数据矩阵,分别为GeneQ、GeneK、GeneV,如公式(1)~(3)所示。这三个数据矩阵可以通过自注意力机制的计算公式进行处理,得到一个处理后的数据矩阵表示,如公式(4)所示。最后,将处理后的数据矩阵与原数据矩阵残差连接,形成一个新的数据矩阵,以进一步提高模型的性能。在本研究中,将3×10的数据矩阵拉平为原始的30个维度,并将其输入分类器部分,以进行分类任务。
分类器部分由三层神经网络组成,包括输入层、隐藏层和输出层。其中,输入层的维度与自注意力模块的输出维度一致,即30 个维度。隐藏层的维度设置为15个,通过降低维度数量,可以减小模型复杂度,提高模型的训练速度和泛化能力。输出层的维度根据癌症分型数量进行设置,以满足多分类的需求。
2.3 模型训练和评价
为了对自注意力机制分类与常见的机器学习分类方法和深度学习方法进行对比,本文选择了决策树、随机森林、支持向量机和逻辑回归等四种机器学习模型,并基于Python 的sklearn 库实现;还选择了循环神经网络并基于Pytorch 实现。此外,本文还分析了去除自注意力模块的SSRNN 的性能以执行消融研究,此方法可称作人工神经网络(artificial neural network,ANN)。
具体地,将样本数据分为训练集和测试集,采用默认参数的方式对上述四种模型进行训练和测试,以比较它们与自注意力机制分类的分类效果和性能表现。
Logistic 回归(logistic regression,LR)是一种基于概率的分类模型,它通过拟合样本的特征和标签之间的关系,预测新样本的类别[29]。决策树(decision tree,DT)是一种常见的分类模型,它通过构建树形结构,根据特征值之间的关系进行分类预测[30]。随机森林(random forest,RF)是一种基于决策树的集成学习方法,它通过组合多个决策树的预测结果,提高分类的准确性和泛化能力[31]。支持向量机(support vector machine,SVM)是一种二分类模型,它通过构建一个最优超平面,将不同类别的样本分开[32]。循环神经网络(recurrent neural network,RNN)是一种用于处理序列数据的神经网络模型。相较于前馈神经网络,RNN 在处理序列数据时具有持续性的特点。
模型的构建是基于Python 的Pytorch 框架实现的。为评估模型的效果,对模型进行了五折交叉验证,随机选择80%的数据作为训练集训练模型,20%的数据作为测试集评估模型效果。选择准确率(accuracy,ACC)、灵敏度(sensitivity,SEN)、精确率(precision,PRE)、F1 得分(F1-score,F1)和受试者工作特征(receiver operating characteristic,ROC)的曲线下面积(area under curve,AUC)等指标对模型效果进行评价。
3 研究结果和讨论
本文研究结果主要包括聚类分析、生存分析、预测性能和生信分析,其中生信分析包括基因表达热图、差异分析和功能富集分析等。
3.1 聚类分析
使用了具有临床信息的514 例肿瘤组转录组数据与生存数据相结合,进行单因素Cox回归分析。本文将过滤条件P值设置为0.001,最终筛选得到了358 个与生存相关的蛋白编码基因。
为了进一步探究KIRC 的亚型分组,使用R 语言的NMF包对这358个蛋白编码基因进行了非负矩阵分解。通过这种方法,可将样本分为不同的亚型,以更好地了解KIRC的异质性。Cophenetic系数图和一致性矩阵如图3 所示,其提示了在不同聚类数条件下Cophenetic 指数的变化情况。Cophenetic 指数越接近于1,聚类效果越好。从图中可以看出,当聚类数k=3 或k=4 时,聚类效果较好。为了确定最终的分型组数,本文结合一致性矩阵图显示的聚类效果进行综合考虑,最终确定分型组数为3。
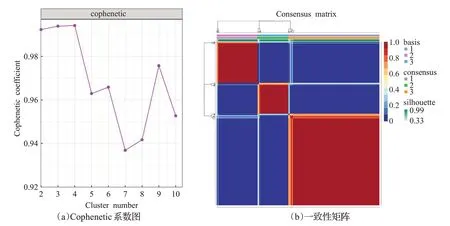
图3 Cophenetic系数图和一致性矩阵Fig.3 Graph of Cophenetic coefficients and consistency matrix
在确定聚类数后,本文利用t-SNE 降维可视化,旨在展示并验证聚类数的合理性,如图4所示,结果表明:当聚类数为3时,样本分类较为合理。

图4 t-SNE降维可视化Fig.4 t-SNE dimensionality reduction visualization
3.2 生存分析
在确定聚类数后,本文进行了各组的临床信息比较和生存分析比较。
一方面,根据样本ID 将转录组数据和临床数据取交集后可获取514 例肿瘤样本,依据亚型分组,将临床信息描述如表1所示。

表1 肿瘤样本临床信息Table 1 Clinical information of tumor samples
另一方面,结合对应的生存信息绘制了生存曲线以对比不同分组之间的生存情况。从生存曲线可以看出,C1、C2和C3组之间存在生存差异(p<0.001),如图5所示。

图5 亚型生存分析Fig.5 Subtype survival analysis
3.3 预测性能
对筛选的358 个生存相关的蛋白编码基因进行了特征筛选。为了选择最优的特征,利用Python的sklearn库选择卡方值前100 个基因作为最终输入模型的特征。接着,使用测试集对模型效果进行了评价,并将SSRNN、去除自注意力模块的SSRNN(简称为ANN)、循环神经网络(recurrent neural network,RNN)和随机森林、决策树、支持向量机、逻辑回归等机器学习分类模型的结果进行了比较。使用五折交叉验证的方法以评估模型的性能,并将5 次结果的均值和标准差展示于表2中。
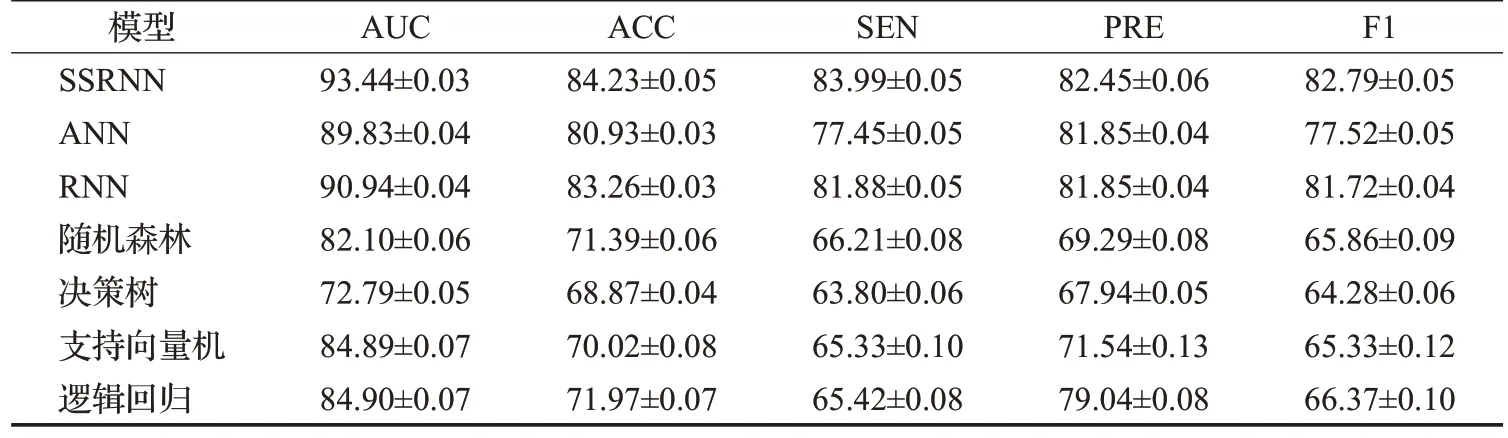
表2 模型效果评估Table 2 Evaluation of model effect 单位:%
在五折交叉验证中,并选择其中表现最佳的一次实验结果来绘制ROC 曲线,各模型的ROC 曲线比较如图6 所示,展示了7 种方法五折交叉验证中最佳一次的结果。综合分析结果,发现自注意力分类模型相对于其他分类方法取得了更好的分类效果。
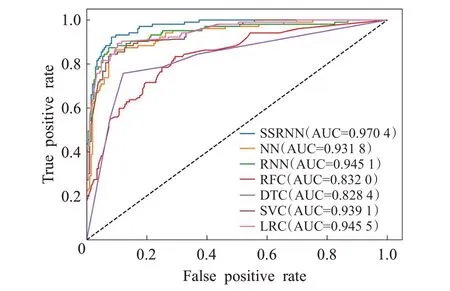
图6 ROC曲线图Fig.6 ROC curve
3.4 生信分析
基于聚类获取的三种亚型执行生信分析,包括基因表达热图、差异分析和功能富集分析等步骤。
首先,对三种亚型绘制肿瘤差异基因的表达热图,如图7 所示。结果表明:三种亚型的基因表达存在差异。C3的基因大多数处于低表达状态,而C1和C2的基因不仅存在高表达而且存在低表达,且在高表达部分的基因存在差异。结合三种亚型的生存曲线,C3 的预后生存最佳,推测基因的低表达指示较好的生存预后。
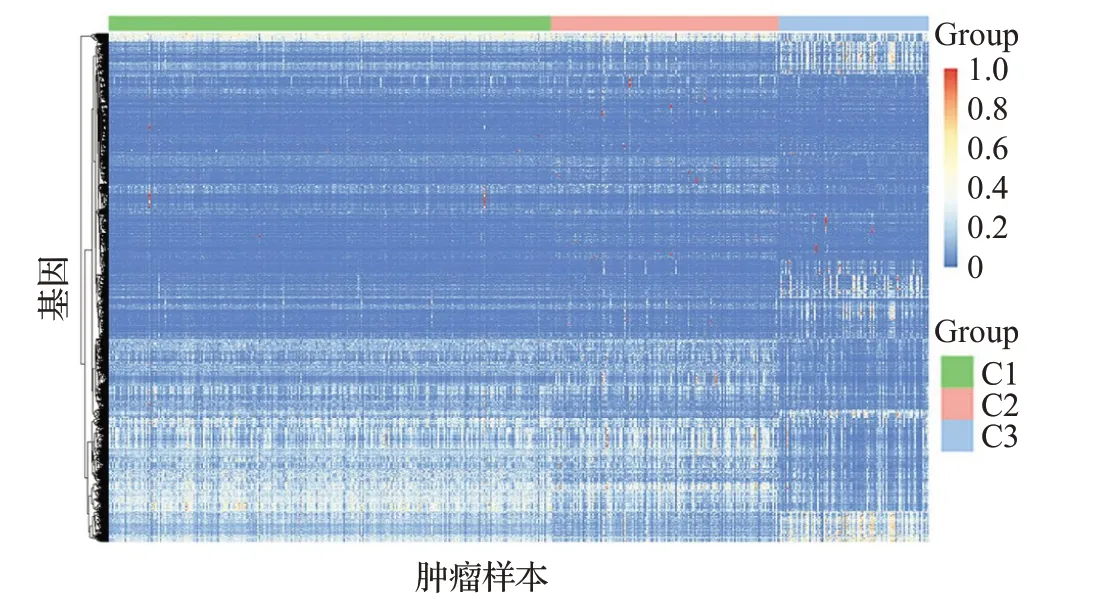
图7 肿瘤差异基因的表达热图Fig.7 Heat map of differential gene expression in tumor
其次,对三种亚型执行差异分析,如图8 所示。将差异倍数设置为1,校正P值设置为0.05。结果表明:对三种亚型两两间进行差异分析并绘制火山图,差异基因取交集,共可获取266个差异基因。这些基因的差异表达可能与肿瘤的发生、发展等方面有关。

图8 差异分析火山图Fig.8 Difference analysis volcano map
最后,使用Metascape 分析工具[33]对从组学数据中筛出的差异基因进行功能富集分析,基于gene ontology(GO)和kyoto encyclopedia of genes and genomes(KEGG)两大基因功能注释数据库选择路径,旨在聚集相似功能的基因并关联生物学表型。两种路径富集的结果如图9所示,均展示前20个富集结果。各气泡表示一个通路或功能项,气泡的大小表示富集分析的显著性程度,颜色表示富集分析结果的不同分类。从整个集群中选择了一个具有代表性功能的子集,并将其呈现为节点图,如图10 所示。节点代表富集的功能。节点大小取决于具有此功能的基因数量,数量越多则节点越大。相似度得分>0.3的功能通过边缘连接,相似度得分越高则边缘越粗。以节点颜色代表集群ID,即相同颜色节点属于同一集群,具有相同网络集群ID 的节点通常彼此靠近。
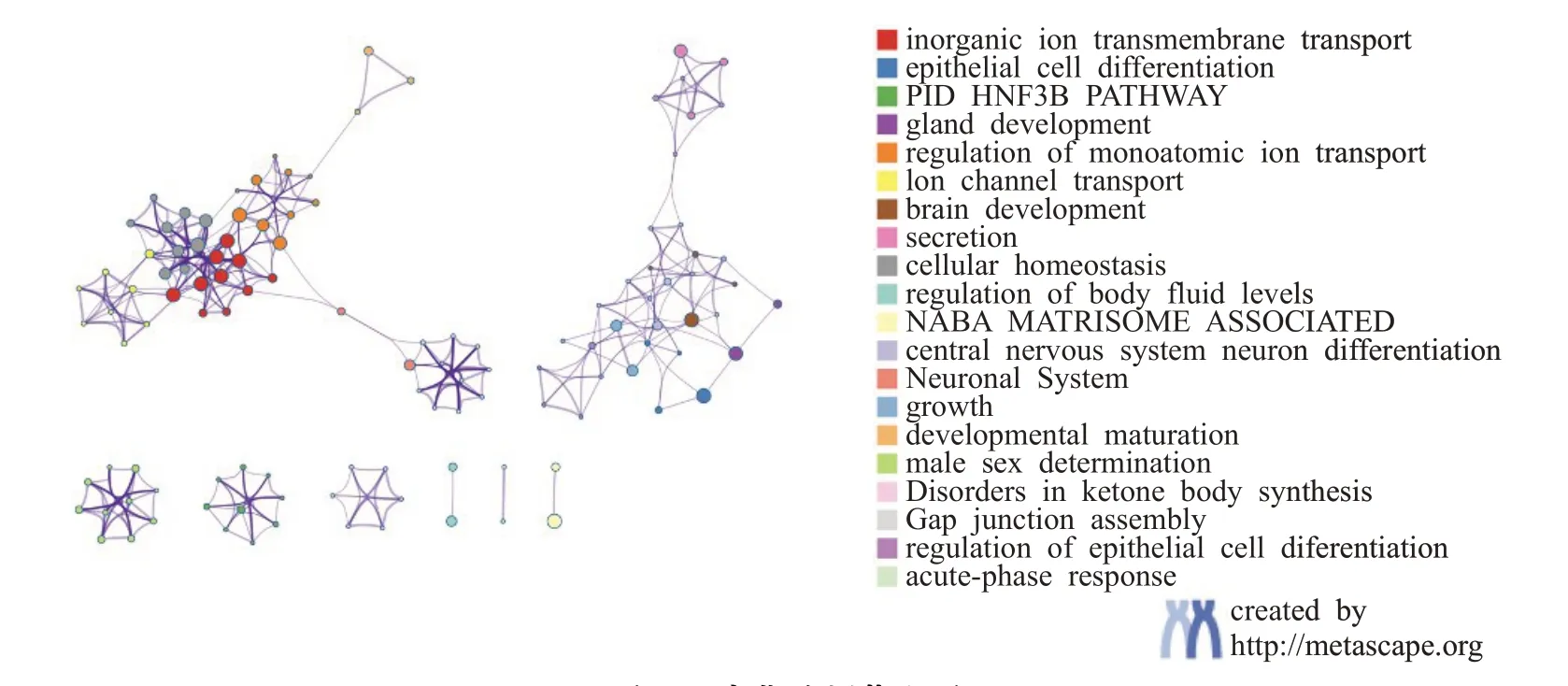
图10 富集分析节点图Fig.10 Enrichment analysis node diagram
3.5 讨论
在确定并筛选出了与癌症发生相关的差异基因后,本文进行了两类富集分析。GO分析提示基因功能主要集中于被动跨膜转运蛋白活性、通道活性、受体配体活性、信号受体激活剂活性和离子通道活性等,而KEGG分析提示通路主要集中于神经活性配体-受体相互作用、细胞因子间受体的相互作用、钙信号通路和细胞粘附分子等。这些生信分析结果揭示了KIRC细胞在跨膜转运等细胞活动中的异常性,并提示了与癌症进展相关的通路。
在聚类分析中,本文结合生存信息筛选出了与生存相关的358 个基因,并对这些基因进行了非负矩阵分解,确定了KIRC 的三种亚型。由筛选出的基因确定的KIRC的三种亚型在生存时间上存在差异,其中亚型C3的生存预后最佳,亚型C1 的生存预后次之,亚型C2 的生存预后最差。因此,本文有效地区分了三种亚型,填补了相关领域的空白,可将三种亚型各自命名为高危型、中危型和低危型。这种亚型划分体现了本文方法和结果的创新,有利于进一步深入探究癌症亚型分类具有重要的理论和实践意义,为癌症的精准治疗提供了有力的支持。
在既往研究中,随着组学数据收集技术和人工智能分析技术的进展,以自编码器(autoencoder,AE)架构为代表的深度学习组学分析方法被广泛用于癌症亚型的识别[34-35]。Tong等[36]提出了ConcatAE方法,该方法连接了从组学数据中学到的特征。Yang 等[37]提出了Subtype-GAN方法,该方法使用单独的多输入多输出神经网络来建模多组学数据。虽然这些方法在癌症亚型识别方面表现出了良好的性能,但它们忽略了数据间的内在关系,可能会丢失部分有意义的信息。
本文预测性能结果揭示,SSRNN 可有效地区分癌症亚型,在各个评价标准上都取得了最佳的效果这进一步证实了SSRNN在癌症研究中的重要性和实用性。在去除自注意力机制模块后,模型效果大幅度降低,这证实了自注意力机制的重要性。自注意力机制的优势[38-40]可总结如下:(1)有利于获取数据间的相关性,提高对重要信息的利用,进而提高模型效果。(2)可更好地提取特征,避免特征之间的冗余和噪声,从而提高模型的准确性和泛化能力。(3)可根据输入数据的不同特征和重要性自动地进行权重分配和特征提取,能够更好地捕捉数据之间的关系,从而有效地提高模型的分类能力。(4)采用了多层神经网络的组合,可以更好地处理非线性分类问题,适用于更复杂的数据集和分类任务。(5)具有较强的准确性和稳健性,可对数据进行更深入的分析和理解,从而更好地指导实际应用。
此外,SSRNN的应用将有助于更好地理解KIRC的发病机制,并为其治疗提供新的思路和方向。本文对基于聚类获取的三种亚型执行生信分析,包括基因表达热图、差异分析和功能富集分析等步骤。这些生信分析结果为深入研究差异基因的功能和通路提供了重要的参考,有助于从生物学角度解释肿瘤发生和发展的机制。一方面,生信分析有利于验证聚类分析的合理性。另一方面,研究人员可根据SSRNN 获得的生物标志物对癌症进行更多的亚型分析研究,为临床研究提供宝贵参考。
基于自注意力机制构建的分类模型已经取得了较好的分类结果,但仍存在一些局限性。首先,本研究仅针对KIRC进行探究,需要扩展到其他癌症分型。其次,模型构建基于TCGA数据划分的训练集和测试集,缺少外部数据验证模型可靠性和泛化性。第三,数据仅考虑KIRC 蛋白编码基因的单一组学数据,需探究多组学数据结合以提高分类效果和精度。未来,将探究其他癌症分型,将基因分型与临床结局进行对应分析,结合多组学数据,并优化模型结构和参数以提高效果和泛化性。
4 总结
本文纳入了KIRC 的转录组学数据,结合非负矩阵分解的方法确定了亚型分组,并利用自注意力机制构建亚型分类模型,提出了自注意力亚型识别神经网络(SSRNN)。研究结果证实了SSRNN 具有优异的性能表现和分类效果。在实际应用中,可以根据具体的数据集和任务,选择合适的分类模型,以实现更加精确和可靠的分类预测。因此,SSRNN 具有较高的预测精度和稳健性,可有效地利用组学数据进行KIRC的生存预测,可较好地指导临床诊治工作,具有较高的方法学意义和应用价值。
猜你喜欢
小雪花·成长指南(2022年1期)2022-04-09
家庭医学(下半月)(2020年1期)2020-05-11
海峡姐妹(2018年7期)2018-07-27
特别健康(2018年4期)2018-07-03
特别健康(2018年2期)2018-06-29
传媒评论(2017年3期)2017-06-13
第二课堂(课外活动版)(2016年2期)2016-10-21
中国病理生理杂志(2015年8期)2015-12-21
中国当代医药(2015年30期)2015-03-01
癌变·畸变·突变(2014年2期)2014-03-01
