
西达本胺对食管鳞癌细胞的抗肿瘤作用及机制
2023-12-11 07:18高曼棋赵宝生杨文倩唐家萍刘玉珍
南方医科大学学报 2023年11期
高曼棋,赵宝生,杨文倩,4,唐家萍,4,刘玉珍,4
新乡医学院第一附属医院1胸外科,4生命科学研究中心,河南 卫辉 453100;2河南省食管癌转移及转化医学重点实验室,河南 卫辉 453100;3新乡医学院食管癌研究所,河南 卫辉453100
我国是食管癌高发国家,世界上半数以上的食管癌发生在我国[1]。其中最常见的病理类型为食管鳞状细胞癌[2]。尽管近年食管癌治疗技术不断提高,但其5年生存率低于20%[3]。因此,寻找新的食管癌治疗方法从而提高疗效为临床所亟需。组蛋白乙酰化水平由组蛋白乙酰基转移酶(HAT)及组蛋白去乙酰化酶(HDAC)共同调控。HAT催化乙酰辅酶A的乙酰基转移至组蛋白氨基末端的赖氨酸残基上,使组蛋白乙酰化,导致染色质结构松弛,激活基因转录。HDAC使组蛋白去乙酰化,使染色质结构紧密,抑制基因转录,其中包括参与细胞分化、凋亡和细胞周期的基因从而促进肿瘤发生发展[4]。多项研究表明,HDAC在多种血液系统肿瘤及实体瘤中存在高表达现象,且与患者的不良预后相关,通过HDAC抑制剂或siRNA干扰来抑制肿瘤细胞中HDAC可抑制肿瘤细胞的增殖[5,6]。因此,HDAC被认为是抗肿瘤治疗靶点。组蛋白去乙酰化酶抑制剂(HDACI)以HDAC为靶点,通过抑制HDAC,阻止组蛋白去乙酰化,导致组蛋白乙酰化水平增加,进而促进抑癌基因转录,发挥抗肿瘤作用,成为一种新型抗肿瘤药物。西达本胺(CHI)是由我国自主研发的首个HDACI,通过抑制HDAC1、2、3、10亚型抑制细胞增殖[7],目前该药在临床上用于治疗外周T细胞淋巴瘤[8]及联合芳香酶抑制剂治疗乳腺癌[9]。研究表明,西达本胺引起肺癌[10],肝癌[11],胰腺癌[12]实体瘤细胞增殖抑制和凋亡。TCGA数据库显示,与正常组织比,HDAC1、2、3、10在食管癌组织中高表达[13]。研究发现[14],HDACI曲古抑菌素A、伏立诺他抑制食管癌细胞增殖,西达本胺对于食管癌的作用及机制尚不明确。本研究通过体外细胞实验和体内动物实验,探讨西达本胺对食管癌的作用及可能机制,为西达本胺用于食管癌治疗提供理论依据。
1 材料和方法
1.1 试剂
西达本胺(Selleck);细胞培养基RPMI 1640(Corning);胎牛血清(FBS)(CLARK);1%青霉素链霉素混合液及二甲基亚砜(DMSO)(索莱宝);Annexin V FITC/PI 细胞凋亡检测试剂盒(上海生工生物工程);细胞周期与凋亡检测试剂盒、MTT试剂、RIPA 裂解液和ECL 化学发光试剂盒(上海碧云天);p21、cyclin D1、Acetyl-Histone H3(Lys9)(H3K9Ac)、H3、cleaved-PARP、PARP、cleaved caspase-3、caspase-3、p-Akt、Akt、p-ERK1/2、γH2AX、Ki-67(免疫组化用)、CD31 均(CST),Ki-67(Santa),ERK1/2和β-actin 单克隆抗体(武汉三鹰),辣根过氧化物酶标记山羊抗兔IgG(Abcam)。
1.2 细胞系
食管鳞癌细胞系KYSE-150、KYSE-450和KYSE-510细胞购自南京科佰生物科技有限公司,人脐静脉内皮细胞(HUVECs)购自武汉普诺赛生命科技有限公司。细胞所用培养基为RPMI 1640,含10%FBS 和1%青霉素链霉素混合液,放置于37 ℃、饱和湿度下5%CO2的培养箱中培养。
1.3 方法
1.3.1 MTT 检测细胞增殖 取对数生长期的KYSE-150、KYSE-450细胞和KYSE-510细胞,分别以每孔2×103、3×103、2×103细胞的数量接种于96 孔板;西达本胺组中西达本胺浓度分别为5、10、20、40 μmol/L,并设置DMSO对照组,每组5个复孔,置于37 ℃、5%CO2的培养箱中培养48 h后,加入5 mg/mL 的MTT 10 μL/孔,37 ℃培养箱孵育4 h,弃上清加入100 μL DMSO,使用酶标仪于检测吸光度A490nm值;细胞存活率=(实验孔平均A值/对照孔平均A值)×100%;计算西达本胺对上述细胞的半数抑制浓度(IC50)。
1.3.2 克隆形成实验检测细胞克隆形成能力 取对数生长期的KYSE-150、KYSE-450及KYSE-510,以1×103/孔接种于6孔板,设置DMSO对照组,西达本胺组中西达本胺浓度分别为5、10、20、40 μmol/L,置于37 ℃、5%CO2的培养箱中培养48 h后更换为完全培养基,培养10 d后,PBS清洗,4%多聚甲醛固定30 min,PBS再次清洗,结晶紫染色15 min后拍照,克隆形成率=染色细胞数/1000×100%。
1.3.3 流式细胞术检测细胞凋亡 取对数生长的KYSE-450细胞,将1×105个细胞接种于直径35 mm 的培养皿中,细胞贴壁后,对照组加入DMSO,西达本胺组加20 μmol/L的西达本胺,培养48 h后,消化后离心,弃上清,1 mL预冷PBS重悬,再次离心,弃上清,加入500 μL结合缓冲液重悬,加入5 μL Annexin V FITC 和10 μL PI,室温避光孵育15 min,流式细胞仪检测细胞凋亡,利用Flowjo 10.8.1 软件分析。
1.3.4 流式细胞术检测细胞周期 取对数生长的KYSE-450 细胞,将2×105个细胞接种于直径35 mm 的培养皿,细胞贴壁后,对照组加入DMSO,西达本胺组加20 μmol/L的西达本胺,培养48 h后,常规消化细胞,离心后吸除上清,2 mL PBS重悬细胞,离心后吸除上清,加入PBS,滴入无水乙醇,-20 ℃固定24 h 后复温,离心后吸除上清,PBS重悬,离心后避光加入500 μL结合缓冲液重悬、25 μL PI和10 μL RNase,37 ℃避光温浴30 min,流式细胞仪检测细胞周期,利用Modfit软件进行分析。
1.3.5 Western blotting 取对数生长的KYSE-450细胞2.5×105,接种于直径35 mm 培养皿,对照组加入DMSO,西达本胺组加20 μmol/L的西达本胺,培养48 h后提取蛋白,BCA法测蛋白浓度。每组取30 μg蛋白样本进行SDS-PAGE凝胶电泳,电泳后转膜,室温下5%牛奶封闭1 h,然后加入一抗4 ℃过夜,所用一抗分别为:cleaved caspase-3、caspase-3、cleaved PARP、PARP、p21、cyclin D1、p-Akt、Akt、p-ERK1/2、ERK1/2、γH2AX、H3K9ac、H3、Ki-67和β-actin(均1∶1000稀释),TBST 洗膜3 次,每次10 min;加入二抗(1∶8000稀释),室温孵育1 h,TBST 洗膜3 次,每次10 min;浸入ECL化学发光液,用凝胶成像仪成像;利用计算机软件ImageJ 1.8.0 进行灰度值分析。
1.3.6 食管癌裸鼠移植瘤模型 20只6周龄BALB/c 雌性裸鼠购自北京维通利华动物技术有限公司,体质量18±2 g,动物生产许可证号:SCXK(京)2019-0001。20只裸鼠随机分为对照组和西达本胺组,每组10只,所有裸鼠均采用右下肢皮下注射0.2 mL细胞悬液(8×106细胞)制备移植瘤模型,第9 天西达本胺组腹腔注射20 mg/kg西达本胺,对照组腹腔注射等体积溶剂玉米油,每3 d给药1次、并称量小鼠体质量及测量肿瘤体积,肿瘤体积=(长×宽2)×0.52,第21天处死小鼠,剥离移植瘤并称重、拍照。动物实验通过新乡医学院第一附属医院伦理委员会审核(伦理号:2020062)。
1.3.7 免疫组织化学染色 取肿瘤组织常规石蜡包埋切片,厚度约为3 μm,按免疫组化检测试剂盒说明书对各组裸鼠肿瘤组织进行免疫组化染色,Ki-67及CD31工作浓度分别为1∶300、1∶200,DAB 显色试剂盒显色,组织变为棕黄色后蒸馏水冲洗后终止显色,苏木素复染切片,晾干,滴加中性树胶,封片,组织切片晾干后,使用尼康H600L光学显微镜观察染色情况,并通过尼康数码相机DS-Fi1c采集图像。
1.3.8 小管形成实验 将Matrigel基质、96孔板和移液枪尖端在4 ℃下完全预冷过夜,96孔板内加入50 μL基质胶将板置于37 ℃培养箱中30 min。取对数生长期的HUVECs,将2×104细胞加入96孔板内,对照组HUVECs加入DMSO,西达本胺组HUVECs加入20 μmol/L的西达本胺,3 h后使用倒置显微镜观察管状结构后进行拍照,利用计算机软件ImageJ 1.8.0计算分支点的数量。
1.4 统计学分析
采用SPSS 27.0软件进行数据处理,数据以均数±标准差表示,两组数据比较采用Studentt检验,P<0.05认为差异具有统计学意义。所有的实验都是独立重复3次。
2 结果
2.1 西达本胺抑制食管癌细胞增殖
不同浓度的西达本胺处理食管癌细胞48 h后,随着浓度的增加细胞存活率下降(P<0.05,图1)。西达本胺处理KYSE-150、KYSE-450、KYSE-510后的IC50分别为32.62、18.21、14.24 μmol/L,KYSE-150、KYSE-450、KYSE-510对西达本胺药物的敏感性依次升高。

图1 MTT检测西达本胺对KYSE-150、KYSE-450和KYSE-510细胞增殖的影响Fig.1 Effects of chidamide on KYSE-150,KYSE-450 and KYSE-510 cell proliferation detected by MTT assay.*P<0.05 vs control group.
2.2 西达本胺抑制食管癌细胞的克隆形成
随着西达本胺浓度的增加,KYSE-150、KYSE-450、KYSE-510细胞形成的克隆集落体积减小,数目减少,克隆形成率随浓度增加而减少(P<0.05,图2)。

图2 西达本胺抑制KYSE-150、KYSE-450和KYSE-510细胞的克隆形成Fig.2 Inhibitory effect of chidamide on colony formation of KYSE-150,KYSE-450 and KYSE-510 cells.*P<0.05,**P<0.01 vs control group.
2.3 西达本胺促进食管癌细胞凋亡
KYSE-450细胞经20 μmol/L西达本胺作用48 h后,相对于对照组,凋亡率增加(P<0.01,图3A);Western blot结果显示,西达本胺上调cleaved caspase-3 和cleaved PARP(均P<0.05,图3B)。
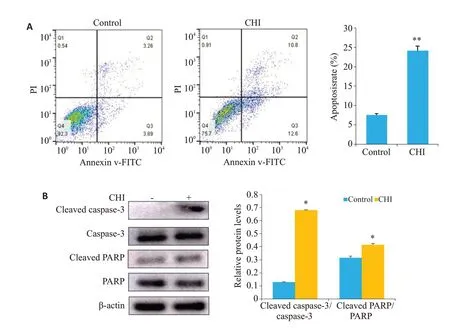
图3 西达本胺处理48 h后对KYSE-450细胞凋亡的影响Fig.3 Effect of chidamide treatment for 48 h on KYSE-450 cell apoptosis.A:Flow cytometry for analyzing apoptosis of KYSE-450 cells.B:Western blotting for detecting expressions of the proteins regulating apoptosis in KYSE-450 cells.*P<0.05,**P<0.01 vs control group.
2.4 西达本胺阻滞食管癌细胞周期
KYSE-450 细胞经20 μmol/L 西达本胺作用48 h后,与对照组相比,西达本胺组G0/G1期细胞比例增加(P<0.01,图4A),S期细胞比例减少(P<0.01),G2/M期细胞比例未见明显变化。结果表明西达本胺使KYSE-450细胞周期阻滞在G0/G1期;Western blot结果显示,与对照组相比,p21蛋白表达上调(P<0.01),cyclin D1表达下调(P<0.05,图4B)。
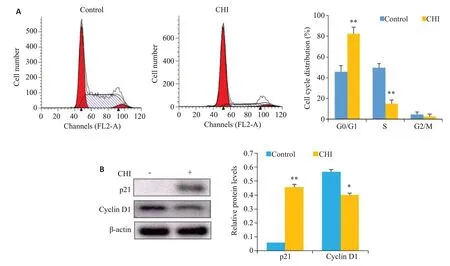
图4 西达本胺处理48 h后对KYSE-450细胞周期的影响Fig.4 Effect of chidamide treatment for 48 h on cell cycle in KYSE-450 cells.A: Flow cytometric analysis of cell cycle of KYSE-450 cells.B: Western blotting for detecting expressions of the proteins that regulate cell apoptosis.*P<0.05,**P<0.01 vs control group.
2.5 西达本胺对通路相关蛋白的影响
KYSE-450 细胞经20 μmol/L 西达本胺作用48 h后,与对照组相比p-Akt水平下调、p-ERK1/2水平下调、γH2AX 水平上调(均P<0.05),H3K9ac 水平上调(P<0.01,图5)。
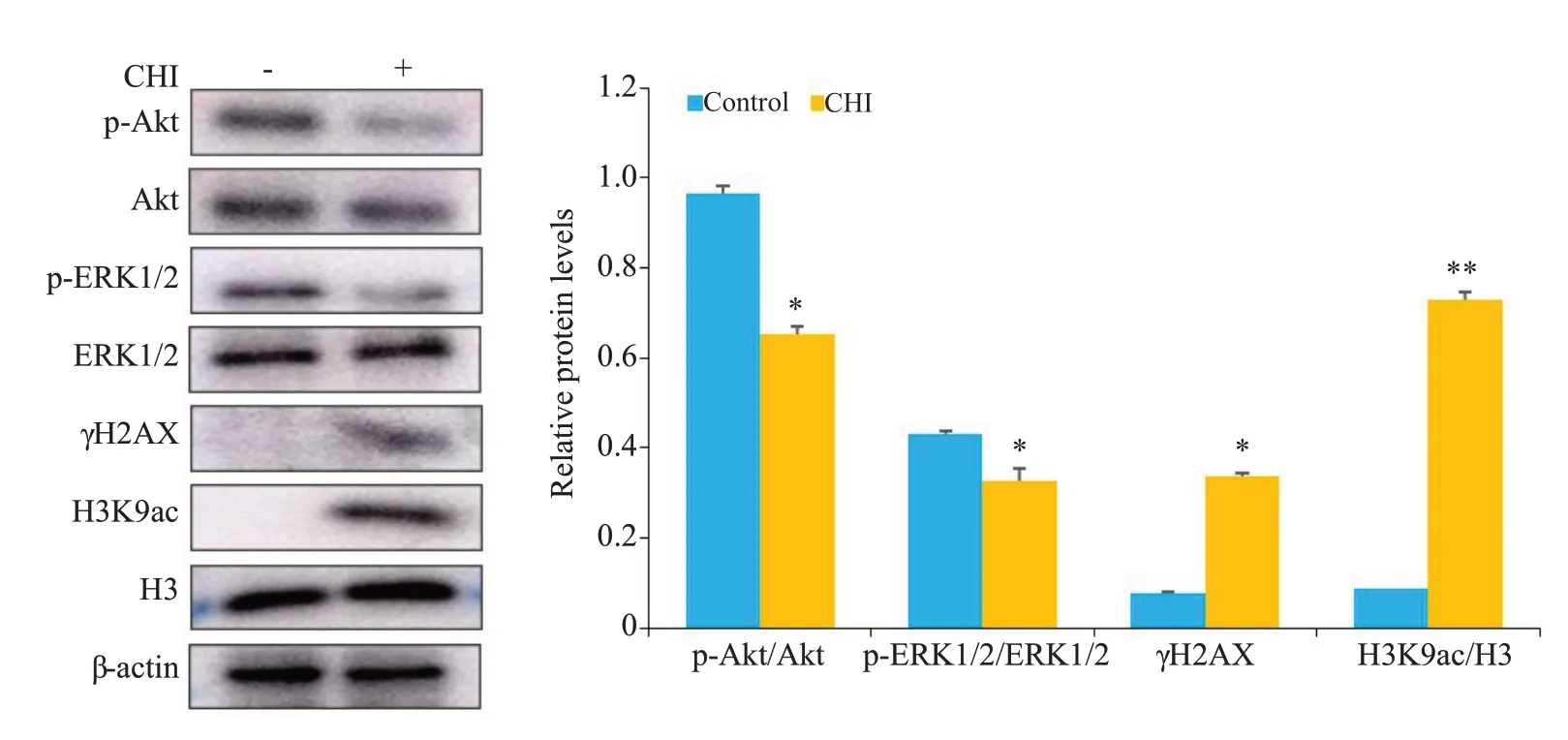
图5 西达本胺对KYSE-450细胞中p-Akt和p-ERK1/2、γH2AX、H3K9ac的影响Fig.5 Effect of chidamide on p-Akt,p-ERK1/2,γH2AX and H3K9ac expressions in KYSE-450 cells.*P<0.05,**P<0.01 vs control group.
2.6 西达本胺抑制食管癌细胞裸鼠皮下成瘤
与对照组相比,西达本胺组裸鼠体重无明显变化(图6A);与对照组相比,西达本胺组肿瘤体积明显减小(P<0.05,图6B);西达本胺组肿瘤标本大小小于对照组(图6C);西达本胺组较对照组瘤重体质量比显著降低(P<0.01,图6D)。通过免疫组化检测各组肿瘤组织中Ki-67(细胞增殖指数)与CD31(血管内皮标志物)的表达。使用积分光密度(IOD)表示Ki-67表达量,结果显示,西达本胺组Ki-67表达量小于对照组(P<0.01,图6E、F)。CD31阳性表达量通过计算微血管密度(MVD)表示,西达本胺微血管密度较对照组减少(P<0.01,图6G、H)。

图6 西达本胺对KYSE-450细胞裸鼠皮下移植瘤的影响Fig.6 Effect of chidamide on subcutaneous KYSE-450 cell xenografts in nude mice.A:Effect of chidamide on body weight of the nude mice (n=6).B: Effect of chidamide on tumor volume in the nude mice (n=6).C:Comparison of tumor specimens from the nude mice(n=6).D:Effect of chidamide on tumor to body weight ratio in the nude mice (n=6).E: Immunohistochemistry for detecting Ki-67 expression in the tumors (n=6,scale bar=100 μm).F:Integrated optical density in the two groups.G:Immunohistochemistry for detecting CD31 expression in the tumors(n=6,scale bar=100 μm).H:Comparison of microvessel density between the two groups.*P<0.05,**P<0.01 vs control group.
2.7 西达本胺抑制细胞增殖及小管形成
KYSE-450 细胞经20 μmol/L 西达本胺作用48 h后,与对照组相比,西达本胺组Ki-67表达水平小于对照组(P<0.01,图7A)。使用HUVECs体外小管形成实验作为体外血管生成研究的模型,使用DMSO和20 μmol/L西达本胺处理HUVECs 3小时,观察小管形成并拍照,与对照组相比,西达本胺组显著抑制HUVECs的成管能力(P<0.05,图7B)。
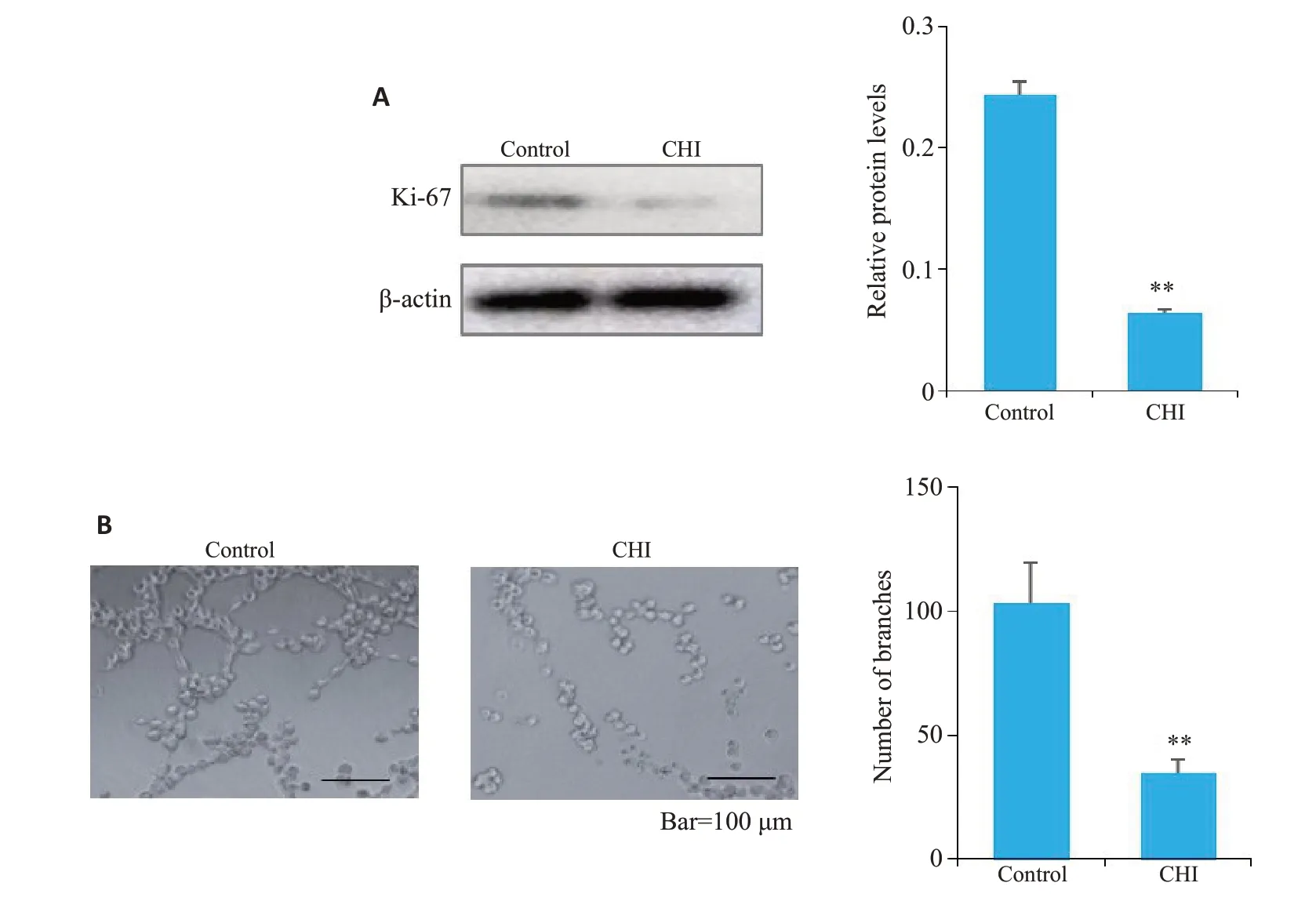
图7 西达本胺对KYSE-450细胞中Ki-67的表达及HUVECs小管形成的影响Fig.7 Effects of chidamide on Ki-67 expression in KYSE-450 cells(A)and tubule formation in HUVECs(B,scale bar=100 μm).**P<0.01 vs control group.
3 讨论
本研究通过MTT实验及克隆形成实验发现,西达本胺呈浓度依赖性抑制食管癌细胞增殖,在KYSE-450细胞中,西达本胺给药48 h的IC50是18.21 μmol/L,其对西达本胺的敏感性低于KYSE-510、高于KYSE-150。因此,本研究选用KYSE-450细胞以及20 μmol/L西达本胺进行后续实验。本研究通过流式细胞术检测西达本胺处理48 h后的食管癌细胞周期和凋亡的情况,结果表明G0/G1期细胞比例和细胞凋亡数增加,说明西达本胺通过阻滞细胞周期G0/G1期及促进细胞凋亡。细胞周期及凋亡对肿瘤的发生发展起着重要作用,细胞周期蛋白依赖性激酶(CDK)抑制因子p21在细胞周期进程起到抑制作用,与CDK复合体结合从而抑制激酶活性阻滞细胞周期进程[15];Cyclin D1可正性调节细胞周期,可与CDKs结合后形成激酶复合物磷酸化视网膜细胞瘤基因(rRb)蛋白,释放出转录因子E2F而促使细胞从G1期转化至S期,促进肿瘤的发生发展[16]。本研究发现西达本胺上调p21且下调cyclin D1表达水平,说明西达本胺通过调控p21和cyclin D1阻滞食管癌细胞周期。半胱氨酸-天冬氨酸蛋白酶(caspase)家族的连续活化在细胞凋亡过程中起着关键作用,起始caspase与促凋亡信号结合后,激活下游caspases 包括caspase-3、caspase-7。Caspase-3 作为细胞凋亡的关键执行者,激活PARP,诱导细胞凋亡[17]。本研究发现西达本胺上调cleaved caspase-3及cleaved PARP的水平促进食管癌细胞凋亡。我们推测西达本胺通过抑制肿瘤细胞增殖、阻滞细胞周期、促进细胞凋亡,进而抑制食管癌的发展。
相关研究表明HDAC1、HDAC3促进Akt磷酸化,进而激活Akt下游靶点,调控细胞凋亡、细胞生长和细胞周期;而抑制HDAC,则阻止Akt激活,进而抑制细胞增殖[18,19]。本研究发现西达本胺下调p-Akt 水平、增加H3K9乙酰化水平,因此我们推测,西达本胺通过抑制HDAC 活性导致组蛋白乙酰化水平升高,进而抑制PI3K/Akt通路,发挥抗食管癌作用。抑癌基因p53是p21的关键调控因子,Akt磷酸化MDM2(MDM2)导致降解p53,从而抑制p21,解除p21对细胞周期蛋白激酶复合物(cyclin D1-CDK4-CDK6)形成的抑制作用,进而使Rb蛋白磷酸化而失活,最终促进细胞周期[20]。Akt依赖的信号通路刺激cyclin D1,促进细胞从G1期转化至S期[21]。Akt活化可减弱caspase-9的活性,进而抑制下游caspase-3,抑制细胞凋亡[22]。本研究发现西达本胺组p-Akt水平下降,p21表达上调,cyclin D1表达下调,cleaved caspase-3及cleaved PARP水平上升。因此推测西达本胺阻滞细胞周期及促进细胞凋亡与其抑制PI3K/Akt通路相关。
细胞外信号调节激酶(ERK)激活后对细胞增殖、细胞凋亡等多种细胞功能产生影响。ERK1/2激活后活化cyclin D1,后者与CDK4/6结合形成激酶复合物进而磷酸化Rb,使得cyclin E与CDK2结合,从而促进G1期向S期转化[23]。ERK1/2通过线粒体途径抑制细胞凋亡,其激活后,一方面通过促进Bcl-2 的表达,抑制细胞色素c释放,另一方面使得凋亡酶激活因子定位于线粒体膜上抑制其与细胞色素c结合,使caspase-9酶原失活,进而抑制下游效应分子caspase-3,发挥抗凋亡作用[24]。本研究发现,西达本胺降低ERK1/2磷酸化及cyclin D1水平,增加cleaved caspase-3及cleaved PARP水平。因此推测,西达本胺阻滞细胞周期及促进食管癌细胞凋亡可能和其抑制ERK1/2激活相关。
研究报道,西达本胺具有协同化疗药物卡铂诱导DNA损伤反应的作用[25],然而,西达本胺单独用药是否具有引起肿瘤细胞DNA 损伤的作用的报道较少[26]。γH2AX 是DNA 损伤后在DNA 损伤位置的H2AX Ser139 位点发生磷酸化的产物,是一种DNA损伤标志物[27]。DNA损伤引起共济失调毛细血管扩张突变激酶的磷酸化,进而活化p53,上调p21抑制细胞周期蛋白依赖性激酶,从而抑制细胞周期[26];DNA 损伤还可以激活caspase-2,引起下游caspase-3 活化,从而诱导细胞凋亡[28]。本研究显示,西达本胺上调γH2AX水平,说明西达本胺导致食管癌细胞DNA损伤。因此推测,西达本胺抑制食管癌细胞增殖、促进凋亡也可能与其引起DNA损伤反应相关。
本研究通过构建食管癌裸鼠皮下成瘤模型,发现西达本胺显著抑制食管癌皮下成瘤。Ki-67是一种与细胞增殖相关的核抗原,其表达水平与细胞增殖活跃度相关。研究发现,Ki-67 表达水平升高促进肿瘤细胞增殖及侵袭转移[29]。血管形成是促进肿瘤生长和浸润转移的重要的条件。研究发现,HDAC1通过激活PI3K/Akt信号通路增加HIF-1α及VEGF表达,进而促进血管生成[30];HDAC10通过激活ERK1/2增加VEGF-2表达,促进血管生成[31]。CD31为血管内皮细胞的标志物,其在微血管表达水平多用于反映肿瘤新生血管的形成,进而评估肿瘤生长情况。在裸鼠皮下成瘤实验中,发现与对照组比,西达本胺显著抑制肿瘤组织中Ki-67及CD31表达量。以上结果表明,西达本胺通过抑制食管癌细胞增殖及血管生成,从而抑制食管癌细胞皮下成瘤。同时,在体外细胞实验中,西达本胺组中KYSE-450细胞Ki-67表达水平低于对照组,并且通过小管形成实验发现西达本胺抑制HUVECs小管形成。进一步在细胞层面上说明西达本胺抑制食管癌细胞增殖及组织血管形成。
西达本胺具有疗效好、低毒性的特点,常见不良反应有血液学不良反应、胃肠道不良反应。在皮下成瘤实验过程中未发现西达本胺组裸鼠存在腹泻、体重明显下降等不良反应。
综上所述,西达本胺抑制食管鳞癌细胞增殖、诱导细胞凋亡、阻滞细胞周期、抑制血管形成,可能与抑制PI3K/Akt、ERK1/2通路及诱导DNA损伤相关。西达本胺通过抑制食管鳞癌细胞增殖和组织血管生成抑制食管鳞癌细胞皮下成瘤。本研究通过探索西达本胺在食管癌中抗肿瘤作用及机制,为西达本胺在食管癌治疗中的应用提供理论依据。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
国际呼吸杂志(2019年4期)2019-03-12
中华老年多器官疾病杂志(2016年7期)2016-04-28
癌症进展(2016年10期)2016-03-20
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
意林·作文素材(2015年16期)2015-08-26
医学研究杂志(2015年5期)2015-06-10
传奇·传记文学选刊(2015年6期)2015-05-30
中国当代医药(2015年16期)2015-03-01

- 南方医科大学学报的其它文章
- 尿路上皮癌抗原1促进滋养细胞功能及对上皮-间质转化转化的调控作用
- Brain iron deposition increases in the bilateral substantia nigra of patients with medication-overuse headache: a quantitative susceptibility mapping analysis
- 益肺散结丸缓解小鼠癌因性骨骼肌萎缩的作用
- 环状RNA hsa_circ_0006834可作为肝细胞癌患者预后的潜在生物标志物
- 慢性间歇性低氧大鼠肾组织差异蛋白的定量分析:基于TMTPRM技术
- 不同滴度MOG-IgG 抗体在MOG 抗体相关疾病中的诊断和临床意义:一项单中心回顾性研究