
噻吩锚定基团的结构修饰对分子-电极结合的影响
2023-12-08 03:15:08雷永久王旭王治业周疆豪陈海舰梁蕾李云川肖博怀常帅
物理化学学报 2023年11期
雷永久,王旭,王治业,周疆豪,陈海舰,梁蕾,李云川,肖博怀,常帅
武汉科技大学材料与冶金学院,武汉 430081
1 引言
分子电子学研究的最终目的是利用单个分子制备性能各异的单分子电子器件,来实现宏观电子器件的功能。通过制造分子电子器件,推动电子器件小型化发展,有望解决传统硅基半导体行业“摩尔效应”即将失效的问题1。研究者们发现带有特定锚定基团的分子会与金属电极发生特定的价键相互作用,从而使分子连接到金属电极之间,形成金属-分子-金属结2,3。利用单分子结可以实现场效应晶体管、负微分电阻、整流、开关等多种功能4–9。其中,锚定基团是构筑单分子功能器件的必要基础,它作为分子电子学的重要研究内容受到了广泛的关注。
当前,可以作为锚定基团的官能团有吡啶基(―Py)、巯基(―SH)、氨基(―NH2)、噻吩、富勒烯等10–12,不同的锚定基团和电极相互作用有着不同的耦合强度和接触构型。一般来说,良好的锚定基团与纳米电极接触时应具有以下特征:耦合强度大、接触构型稳定、接触电阻小等13,14。通常,锚定基团是影响单分子结电学性质的重要因素,它的电子特性影响着分子前线轨道与金费米能级之间的相对位置。锚定基团的缺(富)电性也决定了整个分子是最低空分子轨道(LUMO)还是最高占据分子轨道(HOMO)更加靠近金费米能级,从而可以确定分子的电荷传输通道15。这一点在一些非共轭分子的分子电荷传输中表现得尤为明显。
噻吩及其衍生物具有良好的电学性质和化学可调性,它可以被用于构筑各种各样的有机半导体材料16,例如:有机太阳能电池材料17、有机发光二极管18以及有机场效应晶体管材料19等。另外,噻吩可以与金电极形成有效连接20,21,从而作为构建单分子结所需要的锚定基团。得益于噻吩的这些独特性质,噻吩有潜力作为有机光电功能材料和分子电子学之间的重要桥梁,扩展构筑单分子器件的分子结构基础。因此,研究含噻吩锚定基团的分子的电学特性具有重要意义。
在本文中,我们采用单分子电导测量技术,研究了以噻吩为锚定基团的三种分子(BT-H、BTHex和BT-Cl,如图1)在金电极之间的成结特性。结果表明,三种分子均可以稳定地连接在两个电极之间,产生高(GH)、低(GL)两种电导态,这两种电导态分别由分子与电极通过Au―π和Au―S连接产生。处于BT-H两端的噻吩4号位的H原子被Cl原子或正己基C6取代时,取代基的电子特性会使分子的HOMO能级发生偏移,从而使Au―S构型下的分子电导发生改变,形成GBT-Hex>GBT-H>GBT-Cl的电导趋势。相对而言,三种分子在Au―π构型下的电导没有发生明显的变化。此外,Au―π和Au―S结合构型出现的相对比例也和取代基有关。这些研究对深入理解噻吩与金电极的相互作用以及噻吩作为锚定基团形成的单分子结的电子传输特性具有指导意义。
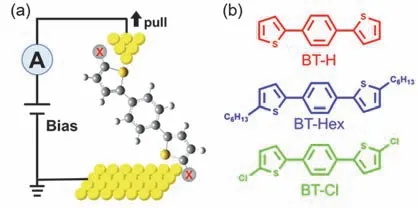
图1 (a) STM-BJ示意图,X代表不同取代基;(b) BT-H、BT-Hex、BT-Cl三种分子的结构Fig.1 (a) Schematic diagram of STM-BJ,X represents substituents; (b) structures of BT-H,BT-Hex and BT-Cl.
2 实验部分
2.1 纳米电极的制备
金基底是通过热蒸发真空镀膜机(型号:Covap,加拿大Angstrom Engineering)在硅片上依次蒸镀20 nm Cr和100 nm Au制成,然后对金基底进行60 s的氢气火焰退火,以获得Au(111)面。
金纳米探针是通过电化学方法腐蚀金丝(99.99%,直径0.2 mm)进行制备,腐蚀液由浓盐酸(36%,国药)和无水乙醇(99.7%,国药)按体积比1 : 1配制而成。制备的金纳米探针在浓硫酸(90%,国药)和过氧化氢(≥ 30%,国药)按3 : 1体积比配制的食人鱼溶液中进行清洗,以去除杂质及残留的有机物,随后用超纯水和无水乙醇对金纳米探针进行多次清洗。
2.2 分子溶液的制备
化学合成的三种分子BT-H、BT-Hex、BT-Cl,结构式如图1b (合成路线见图S1。核磁图可以确定化合物的结构,见图S2–S4,Supporting Information)。将这三种分子分别溶解在1,2,4-三氯苯中(> 99%,美国,Sigma),然后利用超声波清洗器(型号:KQ3200B,昆山市超声仪器有限公司)超声5 min,使其充分溶解,配置成最终浓度为1 mmol·L-1的分子溶液。
2.3 分子电导的测量
我们使用自主搭建的单分子识别仪22–25,采用扫描隧道显微镜裂结(scanning tunneling microscope break junction,STM-BJ)技术采集单分子电学信号。其基本原理是通过步进电机控制金纳米探针靠近金基底,当探针靠近金基底时,利用精度更高的压电陶瓷来控制探针继续接近金基底。当探针与金基底接触融合后,探针以20 nm·s-1的速度远离金基底表面。在提升金针尖的过程中纳米电极之间会形成金原子线,随着探针继续远离金基底,金原子线会断裂并形成纳米间隙。当电极间的纳米间隙与样品分子尺寸相近时,带有锚定基团的分子可以与两个电极连接,形成分子结。随着探针继续远离,分子结会发生断裂。在这个过程中,我们在两电极之间施加100 mV的偏压,在放大器倍数为107V·A-1下以10 kHz的采样率实时记录电极间的电流信号。图1a是分子结的结构示意图。通过控制探针重复接触、远离金基底实现分子结的形成和断裂,我们可以获取上万条电导–距离曲线。通过对数据的统计分析,我们可以得到单分子的电导直方图,并利用高斯拟合得到单分子电导值。本实验在相同的实验条件下对三种分子的电导进行了多次测量,以确保数据的可靠性。此外,我们也对BT-H在放大器倍数106V·A-1下做了单分子电导测量(见图S5,Supporting Information)。
3 结果与讨论
单分子电导测量的电导–距离典型曲线如图2a–c所示。曲线中出现的电导平台证明了分子可以连接到电极之间形成分子结。我们统计了10000多条电导–距离曲线,来绘制一维电导直方图,以此来确定分子的电导峰值。如图2d–f所示,BT-H、BT-Hex和BT-Cl三种分子均出现两个电导峰,这表明三种分子在金电极之间均可以形成两种不同的稳定连接构型。周小顺课题组在苯并二噻吩的单分子电学研究中也检测到了两种电导信号21。我们在近期发表的工作中,解释了以噻吩作为锚定基团形成的单分子结出现两种电导状态的原因25。它们分别是分子的噻吩π轨道和噻吩S原子与金电极发生相互作用产生的。比较三个分子,我们发现通过Au―S作用产生的低电导数值会因为取代基的电子特性不同而发生改变。Cl原子作为吸电子基团,可以通过吸电子效应使噻吩上的S原子电子密度降低,因此减弱S原子与金的结合强度22,26,使BT-Cl的电导相较BT-H降低。而正己基取代基具有微弱的给电子特性,可以增加BT-Hex分子的噻吩硫电子密度,使得BT-Hex的电导比BT-H更高。这一电导顺序GBT-Hex>GBT-H>GBT-Cl也可以通过分子的能级加以解释,即氯原子增大了分子HOMO与Au费米能级之间的差值,而C6链缩小了这个差值(详见图S6,Supporting Information)。
对于Au―π主导的高电导,取代基的影响并不明显。通过计算不同噻吩锚定基团的NICS值27,可以评估分子π电子的离域程度。BT-H、BT-Hex、BT-Cl三种分子对应的NICS(1)ZZ值分别为:-28.08、-24.81、-23.52 (详见表S1,Supporting Information S6)。这三个值比较接近,表明π电子的电子离域程度没有因为取代基的不同而产生明显的差别。这或许可以解释三个分子的Au―π高电导没有发生明显变化的现象。进一步地,我们对BTH、BT-Hex和BT-Cl三种分子进行了几何结构优化,并计算了三种分子与金电极的Au―π相互作用。其中密度泛函方法采用B3LYP,基组对于Cl、C、S和H原子采用6-311G(d,p),Au原子采用LanL2DZ。电极计算模型采用了3个Au原子的Au3团簇,对于分子与电极间相互作用能(ΔE)的计算,我们考虑了色散相互作用28,29(方法采用D3BJ),相关的计算公式如下21:
ΔE=Ecomplex-EAu-cluster-Eorganic-molecule
其中Ecomplex为金与分子通过Au―π结合后的能量,EAu-cluster为金团簇的能量,Eorganic-molecule为分子的能量。结果表明,不同取代基修饰的噻吩与金团簇间相互作用能大小接近(优化后的结构和相互作用能见图3)。这一定程度上可以支持实验中得到的三个分子Au―π电导差别不大的结果。
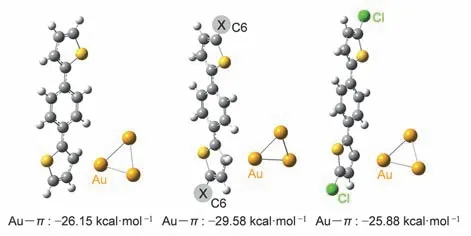
图3 BT-H、BT-Hex、BT-Cl三种分子与金相互作用的优化结构和Au―π的相互作用能Fig.3 Optimized geometries of BT-H,BT-Hex and BT-Cl interacting with gold and the interaction energy of Au―π.
为了进一步研究分子在电极之间的构型,我们将10000多条电导–距离曲线汇总,构建了电导–距离二维强度图(见图4),用来显示电导在不同距离下的分布强度。图中低电导对应的台阶长度分别为0.182 ± 0.07、0.192 ± 0.08、0.185 ± 0.08 nm(统计细节见图S7,Supporting Information)。三个数值十分接近,说明三个分子通过Au―S方式与金电极结合时,分子结的构型相似。高电导对应的台阶长度分别为0.186 ± 0.10、0.141 ± 0.06、0.103 ±0.05 nm,这些长度比Au―S对应的台阶长度更短。文献指出30,Au―π连接是一种非共价相互作用,分子与电极的结合强度相对于Au―S的共价键作用更弱,因此对于相同的骨架长度,Au―π分子结的拉伸长度可能会更短。
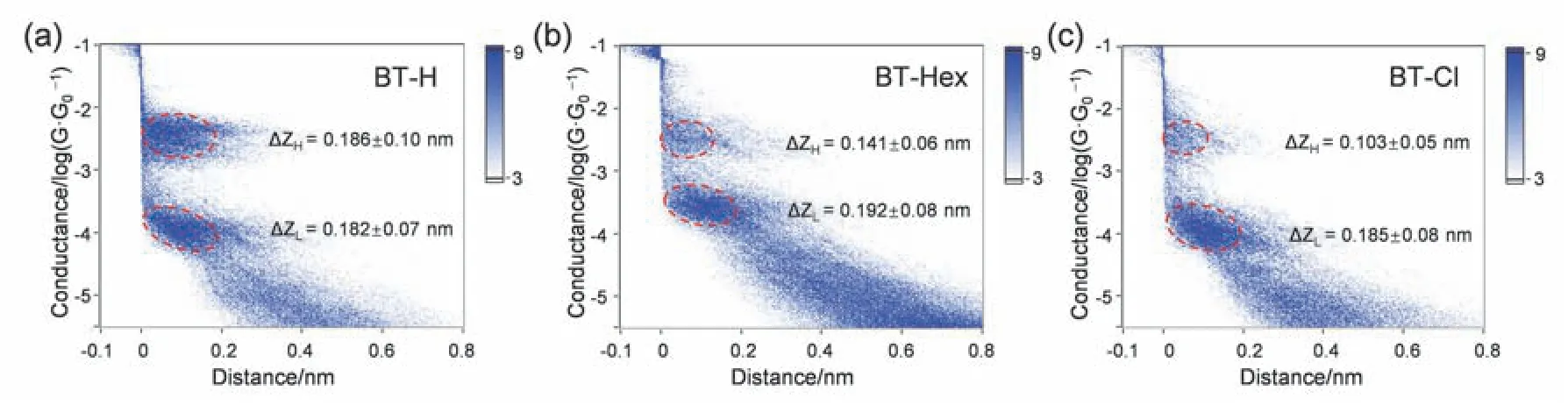
图4 (a–c) BT-H、BT-Hex和BT-Cl三种分子电导–距离二维强度图Fig.4 (a–c) The two-dimensional distribution of molecular conductance–distance traces for BT-H,BT-Hex,and BT-Cl.
进一步地,我们分别对这三种分子的高、低电导做了相关性分析(如图5,详见图S8,Supporting Information),结果表明Au―π和Au―S在同一个拉伸过程中呈负相关31–33。这说明金和噻吩环相互作用时,只会以其中一种构型作为主导。这可能与金电极与噻吩的相对位置有关。
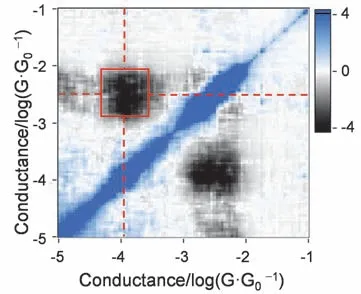
图5 BT-H分子电导的二维协方差直方图Fig.5 2D covariance histogram of BT-H.
接下来,我们对这三种分子的成结率进行了统计分析(见表1)。根据统计结果可知,高电导结合构型(Au―π)对应的成结率随取代基原子半径的增大而降低,这一趋势与我们统计的Au―π分子结长度随取代基原子半径的增大逐渐减小的趋势一致。这可能说明取代基的空间位阻是影响Au―π分子结拉伸长度和成结概率的一个重要因素。
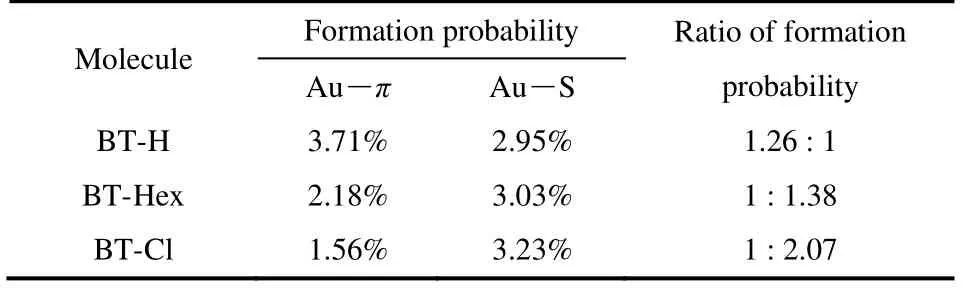
表1 BT-H、BT-Hex和BT-Cl分子两种成结构型的比例Table 1 Formation ratio of two junction configurations for BT-H,BT-Hex and BT-Cl molecules.
4 结论
我们设计并合成了含有噻吩基团的三种分子(BT-H、BT-Hex和BT-Cl),然后利用STM-BJ技术研究了噻吩锚定基团4号位的取代基对分子电导和分子-电极结合构型的影响。研究结果表明,这三种分子在电极之间均存在两种稳定结合构型,分别为Au―π和Au―S结合构型,对应着高电导和低电导状态。H原子被C6或Cl原子取代后,分子的高电导没有受到明显的影响。但由于取代基的电子特性使分子HOMO能级相对于金费米能级发生偏移,使Au―S连接下的低电导发生变化,产生GBT-Hex>GBT-H>GBT-Cl的电导次序。另外,取代基随着原子半径的增加,会导致Au―π结合出现的分子结概率降低,从而影响Au―π与Au―S结合的相对比例。本研究揭示了不同电子性质的取代基对分子结的电荷传输能力以及成结构型的影响,对促进分子器件的发展具有重要意义。
致谢:感谢武汉科技大学材料与冶金学院孙铭骏老师在理论计算上提供的建议。
Supporting Information: available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
猜你喜欢
江苏安全生产(2023年1期)2023-02-08 05:57:50
江苏安全生产(2022年6期)2022-07-29 01:22:32
江苏安全生产(2021年5期)2021-07-16 06:47:16
原子与分子物理学报(2021年1期)2021-03-29 07:29:14
电子制作(2018年14期)2018-08-21 01:38:38
电测与仪表(2016年20期)2016-04-11 11:37:46
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
西南医科大学学报(2016年4期)2016-01-03 01:26:33
应用化工(2014年9期)2014-08-10 14:05:08

- 物理化学学报的其它文章
- 欢迎订阅《大学化学》
- 欢迎订阅《物理化学学报》
- Construction of Z-Scheme MnO2/BiOBr Heterojunction for Photocatalytic Ciprofloxacin Removal and CO2 Reduction
- Holey Graphene for Sodium-Ion Battery Anode Material
- Ir Single Atoms and Clusters Supported on α-MoC as Catalysts for Efficient Hydrogenation of CO2 to CO
- Constructing a CeO2/ZnxCd1-xIn2S4 S-Scheme Hollow Heterostructure for Efficient Photocatalytic H2 Evolution