
利用蔗糖产尿苷二磷酸葡萄糖的大肠杆菌构建
2023-12-08 03:03姚睿赵丽婷陈磊李由然顾正华石贵阳丁重阳
食品与发酵工业 2023年22期
姚睿,赵丽婷,陈磊,李由然,顾正华,石贵阳,丁重阳*
1(江南大学,粮食发酵与食品生物制造国家工程研究中心,江苏 无锡,214122) 2(江南大学,糖化学与生物技术教育部重点实验室,江苏 无锡,214122) 3(江南大学 生物工程学院,江苏 无锡,214122)
尿苷二磷酸葡萄糖(uridine diphosphate glucose,UDP-glucose)是由葡萄糖的半缩醛羟基与尿苷二磷酸的末端磷酸基之间脱水缩合形成的化合物,它不仅是细胞壁合成的前体物质,也是尿苷二磷酸葡萄糖醛酸(uridine diphosphate glucuronic acid,UDP-GlcA)、尿苷二磷酸半乳糖(uridine diphosphate galactose,UDP-Gal)等糖供体的重要前体物质[1-3]。这些核苷酸糖为寡糖、多糖、糖苷等生物活性大分子[4-6]的合成提供多种糖供体,实现了其结构的多样化。UDP-葡萄糖作为其他UDP糖合成的起点,其可用性是高速率合成其他任何相关低聚糖的先决条件[7-9]。但是UDP-葡萄糖价格昂贵且不易获得,因此如何高效获得UDP-葡萄糖成为当下必须解决的问题。
近年来,随着研究者对UDP-葡萄糖研究的不断深入,多级酶联催化反应策略已被广泛用于UDP-糖基供体的循环再生。UDP-糖基供体依赖的糖基转移酶(glycosyltransferase,UGTs)与蔗糖合酶(sucrose synthase, SuSy,EC 2.4.1.13)的偶联反应进行UDP-葡萄糖的循环供应是近年来研究最为广泛的UDP-葡萄糖循环再生体系,它可催化蔗糖和UDP可逆地转化为果糖和UDP-葡萄糖,实现UDP-葡萄糖的一步合成[10]。酶法合成体系虽然能够以廉价的底物合成UDP-葡萄糖,但该催化体系不稳定、操作复杂,往往需要添加辅因子。
随着合成生物学的不断发展,代谢工程逐渐成为一种更为简便和标准化的手段。目前大多数的研究主要集中在以UDP-葡萄糖等核苷酸糖为原料高效合成透明质酸、肝素、柚皮素等[11-13]大分子代谢网络的构建,并未对UDP-葡萄糖从头合成这一关键环节的代谢网络展开深入研究。UDP-葡萄糖作为重要的中间代谢产物,可被快速用于合成含糖基化合物以维持细胞能量,与细胞生长息息相关,这为其检测带来难度。此外,蔗糖合酶的催化底物蔗糖作为丰富的双糖,可使得细胞工厂的构建更加经济和环保,然而目前大多数工业大肠杆菌(Escherichiacoli)不能利用蔗糖[14-16],构建直接利用蔗糖的大肠杆菌细胞工厂,其在蔗糖环境下能否维持渗透压平衡,实现细胞正常生长也是一个难题。
本研究选用遗传背景清晰的E.coliBL21(DE3)为出发菌株,通过引入来自E.coliATCC 13281中非磷酸烯醇式丙酮酸依赖性磷酸转移酶系统(not phosphoenolpyruvate-phosphotransferase system,非PEP-PTS系统)[17-19]中的蔗糖通透酶(sucrose permease, CscB)、果糖激酶(fructokinase, CscK)和来自多型亚硝化螺菌(Nitrosospiramultiformis)的蔗糖合酶(sucrose synthase, SuSy)(图1),以期实现体内的蔗糖到UDP-葡萄糖一步合成,水解产物果糖可以同时被消耗用于细胞生长。构建具有有效利用蔗糖能力的工业菌株将促进蔗糖作为碳源的使用,有助于石油化工经济向生物经济的过渡,为今后利用UDP-葡萄糖生产多种具有不同糖苷键型的葡萄糖基化生物分子提供通用性底盘微生物菌株,也为后续利用UDP-葡萄糖合成相关大分子物质的代谢网络的构建提供了关键的一环。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
本研究所使用的菌株和质粒见表1。

表1 本研究所使用的菌株和质粒Table 1 Strains and plasmids used in this study
1.1.2 实验试剂
蛋白质和酵母粉,Oxoid公司;Na2HPO4、KH2PO4、NH4Cl、MgSO4·7H2O、CaCl2·2H2O、维生素B1、FeCl3·6H2O、CoCl2·2H2O、H3BO3、MnCl2·4H2O、EDTA·2 Na·2H2O、NaCl,国药集团化学试剂有限公司。
UDP-葡萄糖标准品,上海源叶生物科技有限公司;卡那霉素、氯霉素、氨苄青霉素、盐酸大观霉素,Thermo Fisher Scientific公司;异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG)、蔗糖、质粒提取试剂盒、PCR产物回收试剂盒,上海生物工程股份有限公司;引物合成及测序,金维智生物科技有限公司;限制性内切酶、phantaDNA聚合酶,TaKara公司。
1.1.3 培养基
LB培养基(g/L):蛋白胨10,酵母粉5,NaCl 10,将pH值调整至7.0。LB固体培养基另加入1.5%(质量分数)的琼脂粉。
改良的M9蔗糖培养基(g/L):蔗糖20,NaCl 0.5,Na2HPO46.78,KH2PO43,NH4Cl 0.5,MgSO4·7H2O 0.24,CaCl2·2H2O 0.04,维生素B10.001,天冬氨酸2和微量元素1%(体积分数),将pH值调整到7.4。其中蔗糖单独灭菌后再加入M9培养基。
微量元素液(g/L):FeCl3·6H2O 0.83、CoCl2·2H2O 0.01、H3BO30.01、MnCl2·4H2O 0.016、EDTA·2Na·2H2O 6.37。
1.2 实验方法
1.2.1 重组表达质粒的构建
本研究使用的引物如表2所示。选用来自大肠杆菌E.coliATCC 13281中的cscB、cscK基因和来自N.multiformis的susy基因构建蔗糖利用模块。根据NCBI公布的各基因及载体序列设计PCR引物(表2)。通过同源重组构建质粒pACYC-KBS、pET-KBS、pET28a-KBS、SBK-Pcsc-T。基于天然启动子Pcsc构建,选择启动子PT7、PrrnD、Ptac,分别以Pn-F/Pn-R(n为相应启动子)为引物扩增得到启动子PT7、PrrnD、Ptac,通过同源重组连接到经过NheI酶切的载体SBK-Pcsc-T上,得到重组质粒SBK-PT7-Pcsc-T、SBK-Ptac-Pcsc-T、SBK-PrrnD-Pcsc-T。

表2 质粒构建相关引物Table 2 Primers used for plasmid construction
1.2.2 重组菌株在蔗糖-M9培养基中生长特性的测定
蔗糖合酶SuSy在作用的过程中需要UDP的参与,而天冬氨酸是UDP合成的前体物质(图2),在M9蔗糖培养基中补加适量的天冬氨酸替代直接补加UDP得到改良的M9培养基用于发酵。通过改良的M9蔗糖固体平板对重组菌株利用蔗糖的能力进行初步筛选,将平板中生长情况良好的菌株接种到24孔板中,以高通量的形式进行二次筛选。选取性状优良的菌株,在50 mL改良的M9蔗糖培养基中,0 h 时加入0.1 mmol/L IPTG,30 ℃、200 r/min培养,定时检测发酵液OD600nm值监测细胞生长。

图2 尿嘧啶核苷二磷酸合成途径Fig.2 Synthesis of uracil nucleoside diphosphate
1.2.3 CRISPR-Cas9基因编辑系统
基于CRISPR-Cas9系统[20]利用pCas和pTargetF质粒对大肠杆菌的基因组进行编辑。以arsB位点的编辑为例,提取E.coliBL21 (DE3)的基因组DNA,以arsB-A1-F/R、arsB-A2-F/R为引物,扩增得到上下游片段,以质粒SBK-PT7-Pcsc-T为模板,SBK-F/R为引物,扩增得到表达盒,同源重组得到打靶片段SBK-arsB。通过可以靶向arsB的质粒pTar-arsB定向编辑获得重组菌株EP02。重组菌株EP03也以此方法构建。
1.2.4 蔗糖含量的测定
离心收集上清液并用0.22 μm滤膜过滤,通过HPLC测定培养基中残留的蔗糖含量。HPLC检测条件如下:流动相为0.05%的硫酸溶液,流速为0.8 mL/min,示差检测器,液相色谱柱为Dikma CarboPac H+(300 mm×8.0 mm,6 μm),柱温50 ℃,进样量10 μL。
1.2.5 UDP-葡萄糖的生产、提取与分析
离心收集细胞,通过V(乙腈)∶V(甲醇)∶V(水)=2∶2∶1的比例从细胞中提取UDP-葡萄糖。采用由Agilent 1100系列LC系统和AB Sciex API-4000 MS组成的液相色谱-串联质谱(high performance liquid chromatography-tandem mass spectrometry,LC-MS/MS)分析代谢物。
样品在Waters XBridge C-18色谱柱上分离,流速为0.4 mL/min,B为纯乙腈溶液,采用以下梯度:0~0.5 min,50% B;0.5~1 min, B线性上升至90%;1~1.25 min,90% B;1.25~1.5 min,B线性下降至50%;1.5~2 min,50% B,采用多重反应监测(multiple reaction monitoring,MRM)模式获得质谱。用购买的UDP-葡萄糖(纯度>98%)建立校正曲线[21-22]。
1.2.6 荧光定量PCR方法
使用含有目的片段的单拷贝克隆质粒作为标准品,测定标准品浓度,并对其进行10倍稀释,设置7个梯度,以不同浓度的标准品作为模板进行RT-PCR。反应体系:上下游引物各1 μL,SYBR Green Master Mix 25 μL,模板DNA 1 μL,dd H2O 22 μL。以标准品拷贝数的对数作为横坐标,测得的Ct值作为纵坐标,绘制标准曲线。
2 结果与分析
2.1 蔗糖通透酶基因、果糖激酶基因和蔗糖合酶基因的克隆及表达
本研究将合成得到目的基因cscB、cscK和susy克隆到质粒pET28a的EcoR I、NheI两个酶切位点之间,基因大小分别为1 248、915、1 385 bp,测序结果与GenBank报道的基因序列完全一致,表明重组质粒pET28a-cscB、pET28a-cscK、pET28a-susy构建成功。
通过聚丙烯酰胺凝胶电泳验证重组菌株中酶的表达情况,酶CscB(蛋白ID:ABG82031.1)、CscK(蛋白:ID ABG82030.1)和SuSy(蛋白ID:WP_011381564.1)蛋白分子质量分别为45.8、33.6、87.5 kDa。在诱导表达过程中发现菌株ESBK01生长情况较差,其余菌株生长情况良好(图3-A、图3-B),推测过表达蔗糖通透酶CscB可能会对大肠杆菌的生长产生影响。以天然启动子Pcsc低表达CscB[23],结果显示该酶可以正常表达(图3-C),因此在构建UDP-葡萄糖合成途径时应适当下调cscB基因的转录水平。上述结果表明基因cscB、cscK、susy在E.coliBL21(DE3)中成功表达。

A-摇瓶生长对比图;B-重组酶的SDS-PAGE电泳图(M-标准蛋白 Marker;1-空载破碎液;2-ESBK01粗酶液;3-ESBK02粗酶液; 4-ESBK03粗酶液);C-Pcsc下基因cscB的诱导表达 (M-标准蛋白Marker;1-空载破碎液;2-基因cscB粗酶液)图3 cscB、cscK、susy基因的克隆及表达Fig.3 Cloning and SDS-PAGE results of cscB, cscK, and susy genes
2.2 蔗糖利用和UDP-葡萄糖生产模块的构建
2.2.1 转录水平的质粒拷贝数调节
携带不同表达框的重组质粒构建如图4所示,将利用蔗糖产UDP-葡萄糖途径中涉及到的基因构建到不同拷贝数的质粒上(包括质粒pACYCDuet-1、pETDuet-1以及pET28a)从而调控基因剂量,以筛选出能够实现该途径的重组菌株。为了下调基因cscB的转录水平,按照图4所示构建重组质粒。通过菌落PCR进行验证,PCR片段与对应表达元件大小相近,重组菌株ESBK05、ESBK06和ESBK07构建成功。

A-ESBK05;B-ESBK06;C-ESBK07图4 重组菌株ESBK05、ESBK06、ESBK07的构建Fig.4 Construction of the recombinant strains ESBK05, ESBK06, and ESBK07
为了验证不同拷贝数的质粒对蔗糖利用和UDP-葡萄糖合成的影响,将构建好的菌株ESBK05、ESBK06和ESBK07经过筛选后在添加0.5 mmol/L UDP的M9蔗糖培养基中发酵培养,摇瓶发酵结果如表3所示,重组菌株均未生长。

表3 重组菌株ESBK01~07在蔗糖培养基上的生长情况Table 3 Growth of recombinant strain ESBK01-07 on sucrose medium
2.2.2 不同强度的启动子串联调节
基于对启动子元件特性的认识,通过启动子串联策略构建串联启动子能有效提高启动子转录强度。蔗糖合酶SuSy为途径中的关键酶,因此针对SuSy串联启动子从而进一步增强SuSy的表达强度。构建了分别带有PT7-csc、Ptac-csc、PrrnD-csc的重组质粒SBK-PT7-Pcsc-T、SBK-Ptac-Pcsc-T、SBK-PrrnD-Pcsc-T(图5),将其转化至宿主菌并通过菌落PCR进行验证,PCR片段与对应表达元件大小相近,重组菌株ESBK09、ESBK10和ESBK11构建成功。

图5 重组质粒SBK-Pn-Pcsc-T的构建Fig.5 Construction of the recombinant plasmids SBK-Pn-Pcsc-T
重组菌经过筛选后在添加0.5 mmol/L UDP的M9蔗糖培养基中摇瓶培养,结果如表4所示,重组菌株ESBK09生长,而其他重组菌株仍未生长。结果表明相比于天然启动子Pcsc,启动子串联策略提高了蔗糖合酶SuSy的表达强度,由于启动子强度PT7>PrrnD>Ptac,只有在强启动子PT7介导下的重组菌株才能实现细胞生长,蔗糖利用和UDP-葡萄糖合成途径构建成功。
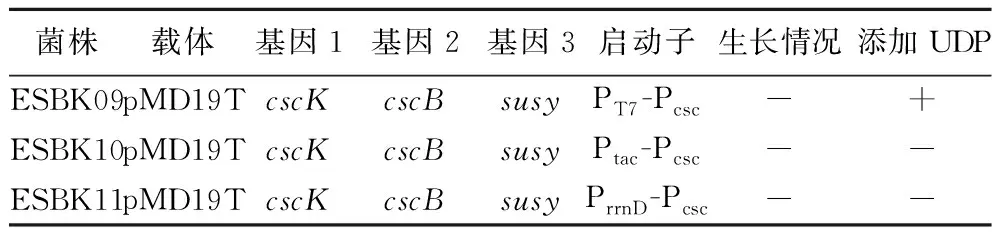
表4 重组菌株ESBK09、ESBK10和ESBK11在 蔗糖培养基上的生长情况Table 4 Growth of recombinant strain ESBK09, ESBK10, and ESBK11 on sucrose medium
2.3 重组菌株ESBK09的细胞生长和蔗糖利用
如图6-A所示,重组菌株在改良的M9蔗糖固体培养基上出现大小不一的菌落,选择生长情况较好的菌株进行实验。原始菌株作为对照组在改良的M9蔗糖培养基中OD600nm值最高仅达到0.5左右,说明原始菌株并不能利用蔗糖。如图6-B所示,重组菌株ESBK09生长情况较好,但延滞期较长,15 h后进入对数生长期,40 h后进入稳定期,OD600值最高可达到6.0左右,生物量可达到5.3 g/L,比生长速率最高可达到0.15 h-1,蔗糖消耗率在60%左右。通过qPCR方法测定重组菌在抗性培养条件下基因拷贝数的变化情况,结果如图6-D所示,重组菌20~40 h进入生长对数期,基因cscK、susy拷贝数最大值出现在25 h左右,基因cscB的最大值出现在20 h左右,基因susy相较于其他两个基因拷贝数明显偏低,蔗糖的消耗以及重组菌株ESBK09的生长进一步证明了该途径构建成功。
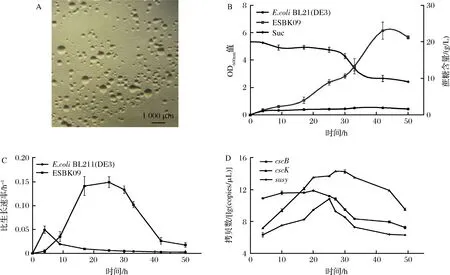
A-M9蔗糖固体平板筛选;B-生长及糖耗曲线;C-比生长速率;D-基因拷贝数图6 重组菌株ESBK09液体发酵Fig.6 Liquid fermentation of recombinant strain ESBK09
2.4 染色体组的整合
在质粒上过表达代谢通路相关的酶存在重组菌株延滞期较长以及质粒不稳定等问题,很难达到稳定生产的目的,将基因cscB、cscK、susy整合到基因组上未筛选到能够利用蔗糖的重组菌株,进而通过转入不同游离质粒的方式分别增强susy,cscK的表达,来实现重组菌株的正常生长。导入重组质粒pET28a-cscK、pET28a-susy、susy-PT7-Pcsc-cscK-T后诱导表达,摇瓶发酵结果如表5所示,重组菌株EPT01、EPT02、EPT03均生长。如图7所示,在3株生长的重组菌株中,重组菌株EPT03在改良的蔗糖培养基中生长良好,10 h进入对数生长期,20 h后进入稳定期,OD600值可达到4.8左右,与重组菌株ESBK09相比有所降低,比生长速率最高可到达0.6 h-1,蔗糖消耗率在70%左右;菌株EPT01和EPT02蔗糖利用能力较差,培养70 h其OD600nm值均低于2.0,且后期再无增长,其中菌株EPT01消耗了大约5%的蔗糖,菌株EPT02消耗了接近2%的蔗糖。总体看来,重组菌株EPT03生长情况与重组菌株ESBK09相比延滞期缩短了近1倍,最高比生长速率是菌株ESBK09的4倍,蔗糖消耗率也有所增加,因此认为重组菌株EPT03略优于重组菌株ESBK09。

表5 重组菌株EPT01、EPT02和EPT03在蔗糖 培养基上的生长情况Table 5 Growth of recombinant strain EPT01, EPT02, and EPT03 on sucrose medium

A-重组菌株EPT01、EPT02生长及糖耗曲线;B-重组菌株EPT01、EPT02比生长速率;C-重组菌株EPT03生长及糖耗曲线; D-重组菌株EPT03比生长速率;E-重组菌株生物量检测图7 重组菌株EPT01、EPT02和EPT03的液体发酵Fig.7 Liquid fermentation of recombinant strain EPT01,EPT02, and EPT03
2.5 EPT重组菌株中相关代谢途径的分析
蔗糖代谢相关基因的引入改变了原始菌株原有代谢途径(图1),为了进一步检测基因cscB、cscK、susy的表达以及对重组菌株主要代谢途径相关基因是否产生了影响,通过qPCR方法测定葡萄糖激酶GLK(EC:2.7.1.2)、葡萄糖磷酸变位酶PGM(EC:5.4.2.2)、葡萄糖磷酸异构酶PGI(EC:5.3.1.9)、葡萄糖-1-磷酸尿苷酸转移酶GALU(EC:2.7.7.9)[24]的基因拷贝数发现(图8-A),在重组菌株EPT01、EPT02、EPT03中,基因glk、pgm、pgi、galU的转录并未受到影响。在重组菌株EPT01中,蔗糖合酶SuSy拷贝数过低,因此果糖无法正常合成,菌株生长较差;在重组菌株EPT02中,当蔗糖进入胞内后在蔗糖合酶SuSy作用下合成UDP-葡萄糖和果糖,但是由于基因cscK表达量不够,胞内果糖无法被高效利用,菌株生长较差。重组菌株EPT03相较于其他两株,基因cscB、cscK、susy都处于高表达水平,该菌株生长情况良好。因此,只有当关键途径基因cscK、susy均处于高表达水平时,该途径才可以达到一个高效利用蔗糖状态,综上所述,重组菌株EPT03的构建实现了蔗糖利用和细胞生长,后续将作为底盘工程菌株用于多糖类物质以及糖复合物的合成。

A-重组菌株EPT01-03拷贝数比较;B-重组菌株EPT03拷贝数图8 重组菌株EPT01、EPT02和EPT03基因拷贝数Fig.8 Gene copy number of recombinant strain EPT01, EPT02, and EPT03
测定重组菌EPT03在抗性培养条件下基因拷贝数的变化情况,结果如图8-B所示,重组菌EPT03在10~20 h进入生长对数期,基因cscB拷贝数最大值出现在10 h左右,基因cscK、susy最大拷贝数出现在5 h左右,基因susy拷贝数相较于基因cscK明显偏低。相较于重组菌株ESBK09来说,基因cscB的拷贝数略有下调,但并未对菌株的生长产生影响,且各个基因拷贝数的峰值明显提前,重组菌株EPT03的生长周期相较于重组菌株ESBK09明显缩短,进一步验证了重组菌株EPT03略优于重组菌株ESBK09。
2.6 UDP-葡萄糖的测定
重组菌株EPT03发酵培养,取发酵25 h的样品进行分析测定,结果如图9所示,UDP-葡萄糖出峰时间为0.241 min,与标样的出峰时间一致,UDP-葡萄糖产量为65.65 μg/g千重,由此判断蔗糖利用和UDP-葡萄糖合成途径构建成功。

图9 标样(UDP-葡萄糖)和样品的液相色谱及ESI质谱图Fig.9 Liquid chromatogram and ESI mass spectrum of standard(UDP-glucose)and smple
3 结论与讨论
本研究提出了一种简单有效的方法,以E.coliBL21 (DE3)为出发菌株,通过引入蔗糖合酶(SuSy)、蔗糖通透酶(CscB)和果糖激酶(CscK),设计并构建了一条以蔗糖为底物合成UDP-葡萄糖的新路径。在此基础上,借助系统代谢工程策略,确定了高拷贝数质粒(pMD19-T)和启动子串联策略(Pcsc-PT7)是这3种酶合成UDP-葡萄糖的最优策略。同时,蔗糖的利用是通过染色体整合和质粒构建共同实现的,使无法利用蔗糖的大肠杆菌菌株能够稳定、高效地利用蔗糖。工程菌株EPT03的成功构建实现了从蔗糖到UDP-葡萄糖的一步合成,也为今后利用UDP-葡萄糖生产多种具有不同糖苷键型的葡萄糖基化生物分子提供强大的底盘微生物菌株[2]。
代谢网络中酶的相对表达水平的平衡对于增强代谢网络是至关重要的[25]。只有当关键途径基因cscK、susy均处于高表达水平,而蔗糖通透酶CscB在内源性csc启动子诱导表达时,该途径才可以达到一个高效利用蔗糖的状态。XU等[26]将大肠杆菌中合成脂肪酸的代谢通路分为3个模块,通过对3个模块的转录水平进行组合优化,进一步提高了脂肪酸的产量;ZHAO等[27]通过调节唾液酸合成途径中不同基因的表达强度,优化相关基因的启动子强度,维持了代谢网络的平衡,大大提高了唾液酸的产量。平衡相关基因表达水平常见的手段包括基因剂量水平的质粒拷贝数调节,转录水平的启动子工程等[28-30]。染色体插入位点也可能会影响调控基因的表达,因此需要评估不同插入位点的转录水平,以便在特定菌株中实现最佳生长速率[23]。
UDP-葡萄糖作为生物体中重要的中间代谢产物,其合成与利用始终处于一种动态平衡状态[31],参与合成多种糖类物质以维持细胞能量,其可用性与细胞生长息息相关,往往生成后便被细胞快速利用,因此难以积累[32]。在大肠杆菌中,UDP-葡萄糖代谢分解成UDP-GlcA、UDP-Gal以及海藻糖-6-磷酸等物质,进而合成下游大分子物质。通过对相关代谢途径最终代谢产物的检测,进而对本研究中重组大肠杆菌中的UDP-葡萄糖流向进行代谢流的监测,有利于后续以重组菌株EPT03作为底盘工程菌的UPD-葡萄糖高效利用代谢网络进行调控。
UDP-葡萄糖在代谢网络中属于中间物质,其可用性是代谢途径中下游物质合成的关键限制因素,在确保UDP-葡萄糖合成的基础上强化下游途径可以高效合成目的产物。首先,UDP-葡萄糖合成模块作为合成大分子物质的代谢网络中至关重要的一部分,可为后续的糖基转移反应提供糖供体,只有先实现UDP-葡萄糖合成才有望通过引入下游代谢途径实现目标产物的合成。DE BRUYN等[11]应用代谢工程策略,引入了蔗糖磷酸化酶,有效合成葡萄糖-1-磷酸,进而合成UDP-葡萄糖,又通过引入多功能葡萄糖基转移酶,使该菌株能够高效生产14种酯类物质。YAN等[33]合理调控大肠杆菌代谢网络,提高宿主细胞内UDP-葡萄糖的合成,通过引入花青素生物合成途径基因在大肠杆菌中成功合成了花青素。此外,UDP-葡萄糖的高效合成有利于下游代谢产物的积累,MAO等[34]过表达了pgm基因和galU基因,提高了UDP-葡萄糖合成途径的碳源流量,在此基础上引入下游途径使产物产量提高了8倍。THUAN等[5]在大肠杆菌中过表达诺卡氏菌属来源的pgm基因和大肠杆菌来源的galU基因促进UDP-葡萄糖的合成进而使下游糖苷的产量大大增加。通过微生物细胞工厂理性设计优化代谢网络,对底盘工程菌株的代谢路径进行改造,动态调控生物合成途径,有利于实现代谢流高效流向目标产物的合成方向。
猜你喜欢
河北医学(2021年10期)2021-10-27
广西糖业(2020年3期)2020-09-25
中国临床医学影像杂志(2019年6期)2019-08-27
食品科学(2018年10期)2018-05-23
浙江工业大学学报(2017年5期)2018-01-22
中国糖料(2016年1期)2016-12-01
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
发明与创新(2015年25期)2015-02-27
中国果业信息(2015年11期)2015-01-23
