
乙型肝炎病毒感染性疾病患者肠道菌群结构和多样性分析
2023-12-07 11:49:48赵成燕周欣怡高冉冉韩丹郑嵘炅鲁晓擘
肝脏 2023年11期
赵成燕 周欣怡 高冉冉 韩丹 郑嵘炅 鲁晓擘
乙型肝炎病毒(HBV)属于嗜肝DNA病毒科病毒,其感染成为全球性公共卫生问题。虽然随着乙型肝炎疫苗的普及,我国在HBV感染防治方面已取得巨大成绩,低年龄组人群HBV感染率大幅度下降,但HBV防治形式依然十分严峻。国内仍约有7000万HBV慢性感染者[1]。HBV感染早期无明确诊断,或对感染者进行持续治疗,则部分患者会在感染后10~15年出现肝硬化、肝功能衰竭、甚至肝细胞癌(HCC)等[2]。国内肝硬化和HCC中分别约有77%和84%为HBV感染所致[3]。探究影响HBV感染者肝损伤及恶化的危险因素,对预防HBV感染者肝硬化、肝衰竭、HCC发生具有重要意义。人体肠道中种类繁多的微生物被称为“肠道菌群”。正常情况下,人体肠道菌群参与宿住新陈代谢,维持宿住身体健康。现有研究证实,肠道菌群失调可能与慢性肝损伤[4]、糖尿病[5]、恶性肿瘤[6]等发生有关。赵志方等[7]研究发现,与健康人群相比,慢性乙型肝炎(CHB)患者体内出现普雷沃氏菌属(Prevotella)、劳特氏菌属(Blautia)、瘤胃球菌属(Ruminococcus)、真杆菌属(Eubacterium eligens group)等菌属失调,单形拟杆菌中的Ruminococcus sp.5-1-39BFAA与丙氨酸氨基转移酶(ALT)、超敏C反应蛋白(HsCRP)、国际标准化比率(INR)水平呈显著负相关,Bacteroides uniformis与ALT水平正相关,提示肠道菌群失调可能在肝损伤发生发展中发挥一定作用。但关于不同肝损伤程度的HBV感染者肠道菌群失调的具体表现,目前尚缺乏相关数据报道。本研究采用基因测序法对HBV携带者、乙型肝炎肝硬化(LC)、肝细胞癌(HCC)、慢加急性肝衰竭(ACLF)、自身免疫性肝炎(AIH)、原发性胆汁性肝硬化(PBC)患者肠道菌群进行测定分析,以明确肠道菌群失调在HBV感染肝脏功能损伤发生、发展中的作用机制,从而为HBV者持续干预提供参考。
资料与方法
一、一般资料
选择2020年2月—9月在新疆医科大学第一附属医院肝病专科接受诊治的133例肝病患者为研究对象,其中HBV携带者(HBV组) 23例、LC(LC组) 25例、HCC(HCC组) 38例、ACLF(ACLF组) 16例、AIH (AIH 组)13例、PBC(PBC组)18例。纳入标准:①HBV组和LC组诊断符合《慢性乙型肝炎防治指南(2019年版)》有关标准[3],ACLF组符合《肝衰竭诊治指南(2012年版)》有关标准[8],HCC组符合《中国原发性肝细胞癌放射治疗指南(2020年版)》中有关标准[9],AIH组符合《自身免疫性肝炎诊断和治疗共识( 2015)》诊断标准[10],PBC组符合《对我国炎症性肠病诊断治疗规范的共识意见》中诊断标准[11];②入组前3个月内未使用过抗菌类药物,4周内未使用过益生菌或促胃肠动力药物;③了解研究内容且签署知情协议书。排除标准:①丙型肝炎病毒等其他肝炎病毒感染性疾病;②具有消化道器质性病变或手术操作史;③具有甲状腺功能异常或自身免疫系统疾病;④无糖尿病、脂肪肝代谢性疾病[2]。6组患者基本资料间比较差异无统计学意义(P>0.05,见表1)。本研究符合医学伦理学标准。
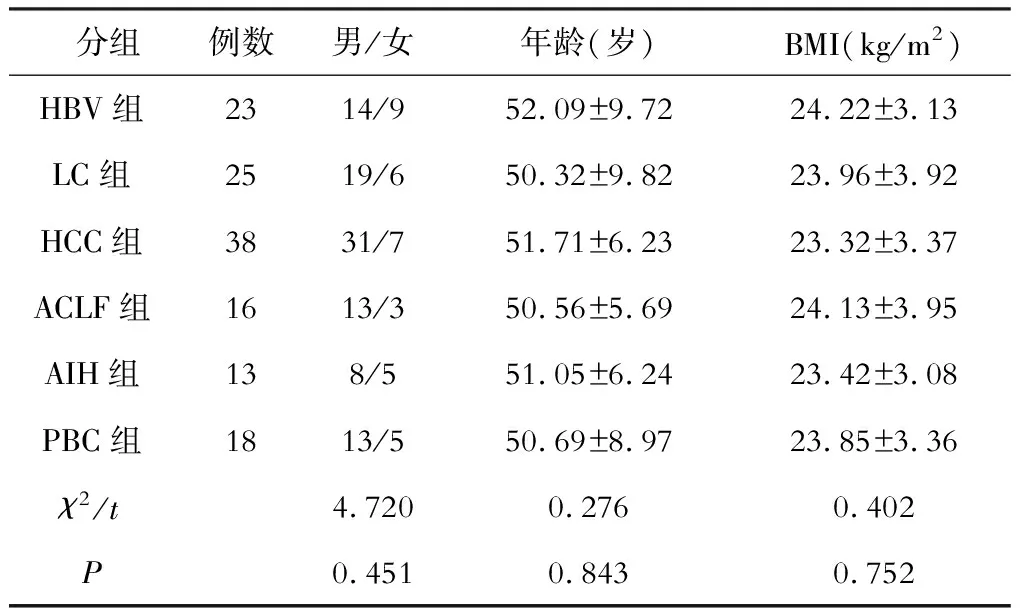
表1 各研究样本基本情况比较
二、研究方法
(一)样本采集 留取纳入者晨起时新鲜粪便样本,采样勺取其中间部分置入采样管,-80 ℃冷藏待用。
(二)DNA提取及扩增 CTAB法(诺禾致源)提取粪便样本中细菌总DNA,1%琼脂凝胶电泳监测所提取总DNA浓度和完整性。无菌水将DNA样本稀释至1 ng/mL。利用特定性引物扩增DNA样本中16S rRNA基因的V3~V4可变区域(PCR扩增条件:98 ℃初变性60 s,随后98 ℃变性10 s,50 ℃退火30 s,72 ℃伸长30 s,共30个循环,之后72 ℃延伸5 min)。PCR扩增产物与相同体积1×负载缓冲液混合,2%琼脂凝胶电泳仪电泳,Qiagen Gel Extraction对混合PCR产物进行纯化。

(四) 生物信息分析 FLASH对剪切的reads进行合并;根据QIIME质量控制流程对原始标签在特定过滤条件下进行质量过滤,得到高质量的洁净标签;UCHIME将标签与参考数据库进行比较,去除嵌合体序列;最终获得有效标签。Usparse软件进行序列分析,相似度≥97%的序列被分配到相同的可操作分类单元(Operational taxonomic units,OTU)中;Silva数据库基于Mothur算法对OTU进行分类信息标注;QIIME软件(Version 1.7.0)计算样本肠道菌群多样性;QIIME软件(Version 1.9.1)计算加权和未加权单株的Beta多样性;R软件(Version 2.15.3)中的ade4包和ggplot2包对原始变量主成分分析(Principal component analysis,PCA)和主坐标分析(Principal Coordinate Analysis,PCoA);QIIME软件(Version 1.9.1)进行非加权算术均值对组法(Unweighted Pair-group Method with Arithmetic Means,UPGMA)聚类。
三、统计学方法
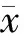
结 果
一、各组患者肠道菌群组成
6组的138个样品共获得83 065条有效数据,经过质控得到69 984条数据,质控有效率为83.83%。按照97%的一致性,将获得有效序列聚类成OTUs,共得到12 022个OTUs。不同组间特有和共有OUT花瓣图分析绘制成韦恩图,其中HBV组独有OTUs数目最多,LC组次之,AIH组最少,见图1。HBV组独有OTUs数目最多,其次为LC组、PBC组、HCC组,AIH和ACLF组最少。

图1 各组肠道微生物OUT分布韦恩图
二、肠道菌群结构
物种注释结果显示,6组样本门水平上最大丰度均处于前10位的分布为Firmicutes、Proteobacteria、Bacteroidota、Actinobacteria、Verrucomicrobiota、Fusobacteriata、Cyanobacteria、Unidentified bacteria、Actinobacteriota、Synergistota,其中以厚壁菌门、变形菌门、拟杆菌门、放线菌门为主,HBV组、LC组、HCC组、ACLF组、AIH组和PBC组样本中最大丰度处于前4位菌门的总占比分布为88.76%、97.22%、96.15%、92.88%、95.91%和93.36%,见图2。

图2 各组门水平物种相对丰度柱状图
在门的水平上,HBV组和HCC组Actinobacteria丰度显著高于LC组,HBV组Actinobacteriota丰度显著高于LC组、HCC组和ACLF组,LC组和ACLF组均显著低于PBC组;LC组Bacteroidota丰度显著高于HCC组;HCC组的Firmicutes丰度显著高于HBV组;同时,HBV组其他菌门丰度均显著高于LC组、HCC组和ACLF组,ACLF组明显低于PBC组,差异均具有统计学意义(P<0.05),见图3。
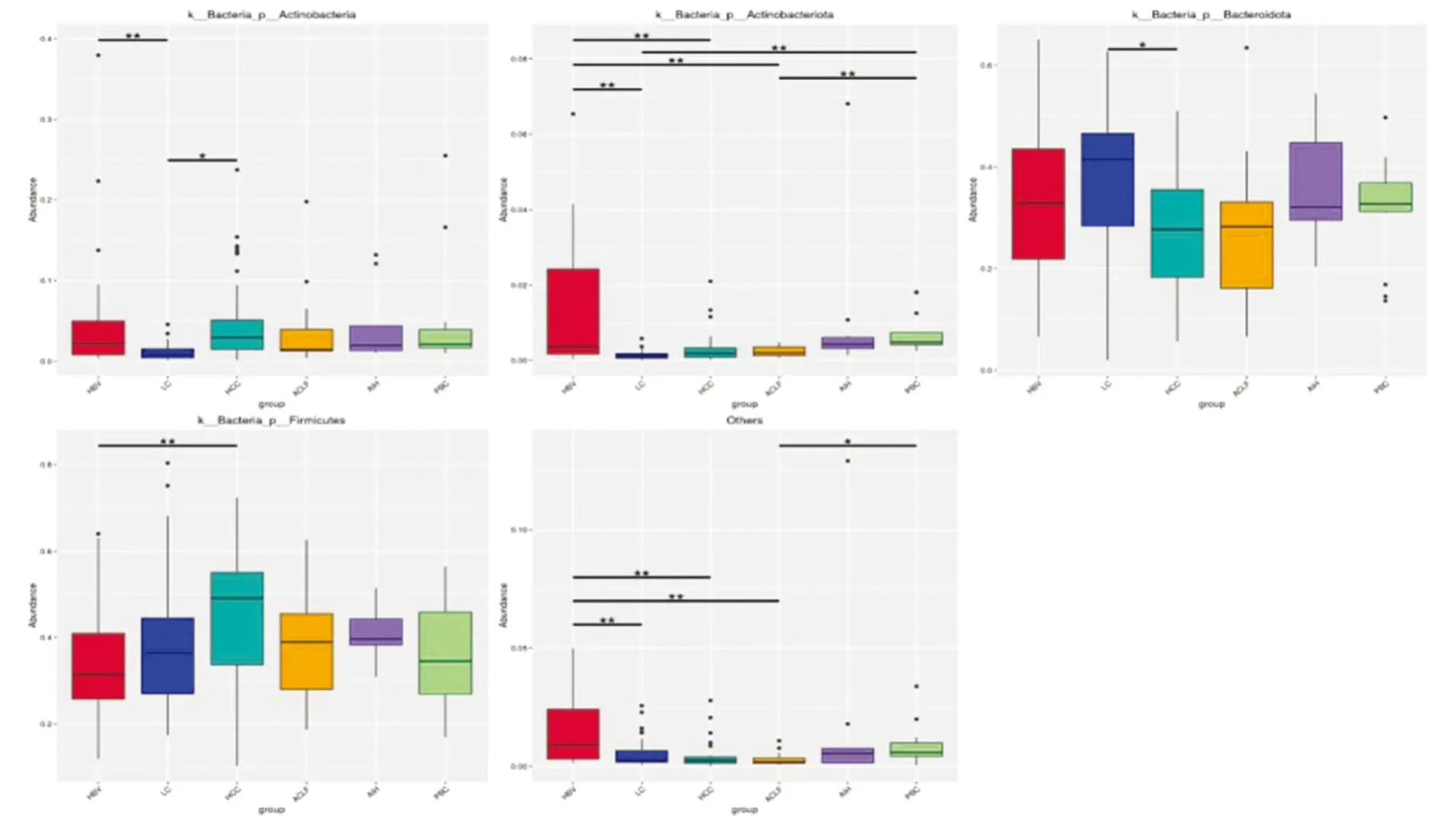
图3 各组门水平上物种显著性差异统计图
三、各组肠道菌清α多样性比较
从每个样本中抽取一定测序量的数据,统计其所代表物种数目,并以此构建稀释曲线,结果显示,随测序量增加,各组观测物种数量不断增加,且最终均趋于平坦,测序数据量和深度均合理(图4左)。Round-Abundance曲线分析显示:HBV组横轴上范围最大,物种丰度最大,PBC组次之,随后依次为LC组、HCC组、ACLF组,AIH组横轴上范围最小,物种丰度最低;纵轴上,HBV组曲线最平缓,物种分布最均匀,其次依次为PBC组、LC组和HCC组,ACLF组和AIH组平缓度相似,两组均匀度相对较差(图4右)。
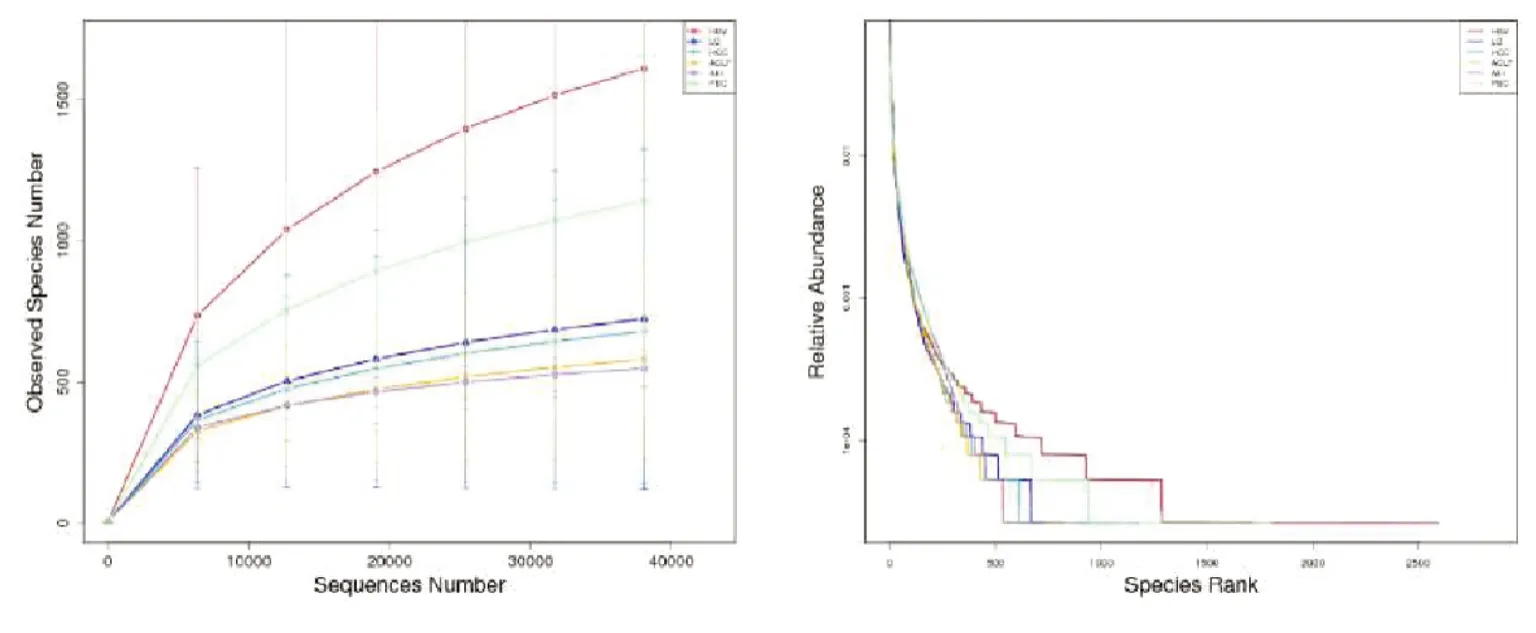
图4 各组样本物种多样性比较(左:稀释曲线;右:Rank-Abundance曲线)
四、各组肠道菌群的β多样性分析
PCA主成分和PCoA主坐标分析显示,6组样本肠道菌群存在明显差异(图A和B)。基于β多样性距离非度量多维尺度分析(NMDS)显示,HBV组、LC组、HCC组和PBC组中样本间离散度较高,组内菌群差异性较大,ACLF组AIH组中样本间距较近,组内群落比较集中。HBV组与LC组、HCC组、PBC组样本多分布于不同区域,组间肠道菌群群落结构差异较大;ACLF组与AIH组样本间距较近,群落结构相似,见图5。
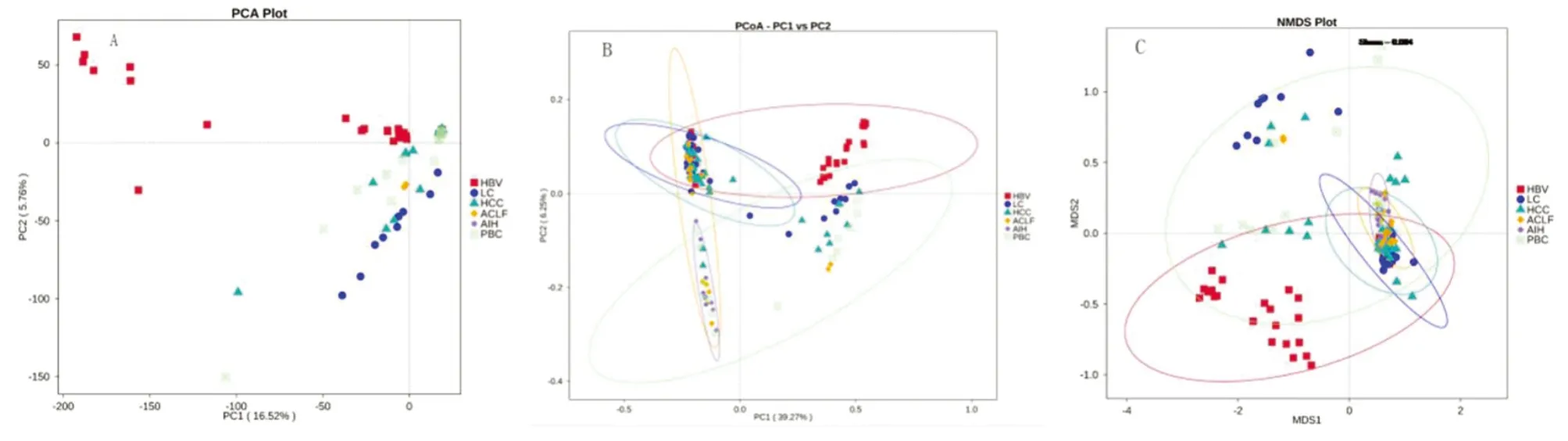
图5 各组肠道菌群多样性的PCA(A)、PCoA(B)和NMDS分析(C)
五、各组样本肠道菌群复杂度分析
HBV组所测物种数明显多于LC组、HCC组和ACLF组,LC组明显低于AIH组和PBC组,HCC组和ACLF组均显著低于PBC组(P<0.05),而AIH组与HBV组、HCC组和ACLF组间所测物种数间比较差异均无统计学意义(P>0.05)(图6左)。HBV组Shannon指数明显高于LC组和ACLF组,LC组、HCC组、ACLF组Shannon指数均明显低于PBC组(P<0.05),但HBV组与HCC组、AIH组与HBV组、HCC组和ACLF组Shannon指数间比较差异均无统计意义(P>0.05)(图6右)。
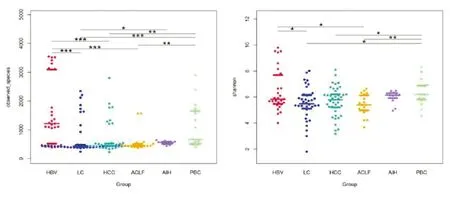
图6 各组肠道菌群复杂度比较
六、各组样本菌群属间差异分析
HBV组样本中属水平菌群含量最丰富的是Unidentified-Chloroplast,丰度最低的为Escherichia-Shigella、Ruminococcus、Pseudomonas、Veillonella;LC组样本中属水平菌群含量最丰富的是Megamonas、Clostridioides、Prevotella-9、Dialister,丰度最低的为Bifidobacterium、Streptococcus、Blautia、[Rumimococcus]-gnavus-group;ACLF组样本中属水平菌群含量最丰富的是Akkermansia、Anaeroglobus、Lactiplantibacillus,丰度最低的为Megaspaera、Ligilactobacillus;HCC组样本中属水平菌群含量最丰富的是Acinetobacter、Ralstonia、Subdologrenulum、Pseudomonas,丰度最低的为Prevotella-9、Parabacteroides;AIH组样本中属水平菌群含量最丰富的是Bacteroides、Blautia、[Rumimococcus]-gnavus-group,丰度最低的Enteroccocus;PBC组样本中属水平菌群含量最丰富的是Prevotella、Alloprevotella、Neisseria、Fusobacterium,丰度最低的Alistipes、Agathobacter,见图7。
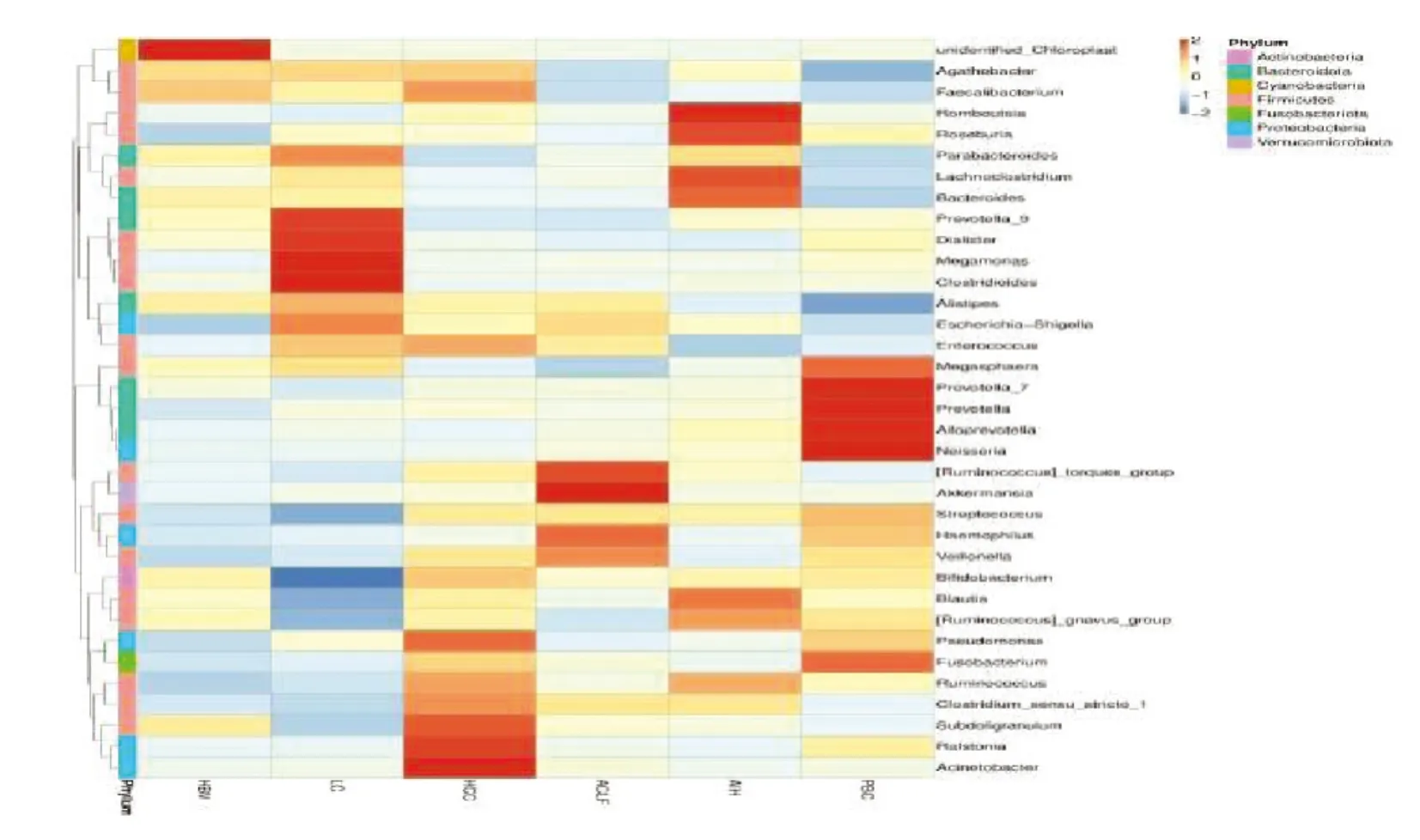
图7 Heatmap图比较差异菌种在各组中的分布
通过对6组样本进行LEfSe分析(LDA阈值设定为4),共发现23个biomarker,其中ACLF组生物多样性最丰富,代表菌群较多,LC组和PBC组次之,随后依次为HBV组和AIH组;HCC组多样性较少,且与其他菌群存在较大重叠,故无差异显著的菌群,见图8。

图8 各组菌群属间菌群LEfSe分析
讨 论
HBV感染所致CHB可引起各种急、慢性肝损伤,进展为LC、ACLF或HCC等,严重损伤人类身心健康,威胁患者生命安全[12]。全球约有2.57亿慢性HBV感染者,88.7万死于CHB并发症[13],38.4万死于LC[14]。肠道菌群是人体最大且最重要的微生态系统,肠道中定植有超过100万亿个微生物,这些微生物参与并影响宿住生理代谢和各种生理病理过程[15-16]。肝病发病机制相关研究发现,慢性肝损伤患者体内会发生肠道菌群失衡,且肠道菌群失衡与HBV感染相关肝病发生、发展有关[17]。Wang等[18]利用Illumina MiSeq测序平台对85例Child-Pugh评分较低的CHB患者肠道微生物组成与22例健康对照者进行比较发现,与健康人群相比,CHB患者肠道菌群发生明显变化,表现为Actinomyces、Clostridium sensu stricto、unclassified Lachnospiraceae、Megamonas4个属OTUs增加,Alistipes、Asaccharobacter、Bacteroides、Butyricimonas、Clostridium IV、Escherichia/Shigella、Parabacteroides、Ruminococcus、unclassified Bacteria、unclassified Clostridiales、Unclassified Coriobacteriaceae、Unclassified Enterobacteriaceae、Unclassified Lachnospiraceae和Unclassified Ruminococcaceae等27菌门减少,这些变化提示肠道菌群失衡与CHB进展具有潜在贡献。
郑婷婷等[19]研究显示,健康人及CHB、LC和ACLF患者肠道菌群均以Firmicutes、Bacteroidota为主,与本研究结果略有不同,本研究以CHB、LC、HCC、ACLF 4个HBV感染不同阶段患者和AIH、PBC患者肠道菌群结构为研究,6组样本中门水平以Firmicutes、Proteobacteria、Bacteroidota为主。这可能与研究对象生活于不同地区有关。李攀[20]研究结果显示,肠道菌群与宿住信息存在广泛联系,地理因素对人体肠道菌群结构和多样性的影响甚至超过了不同地区疾病的影响。邢乐康等[21]研究显示,与健康人群相比,ACLF患者肠道菌群丰度和多样性均下降,且随着肝损伤病情进展肠道菌群失衡现象越严重。6组样本肠道菌群丰度依次为HBV组、PBC组、LC组、HCC组、ACLF组、AIH组,HBV组、LC组、HCC组和PBC组组内菌群差异性较大,组间肠道菌群群落结构差异较大,ACLF组、AIH组中肠道菌群群落比较集中,群落结构相似。证实,HBV感染后随肝脏损伤病情加重,其肠道菌群多样性明显下降,群落结构差异性增大,且相同肝损伤阶段患者体内肠道菌群存在明显个体差异。此外,HBV组和HCC组Actinobacteria丰度显著高于LC组,HBV组Actinobacteriota显著高于LC组、HCC组和ACLF组,LC组和ACLF组的Actinobacteriota及显著低于PBC组;LC组Bacteroidota丰度显著高于HCC组;HCC组的Firmicutes丰度显著高于HBV组。Actinobacteria和Actinobacteriota下的双歧杆菌科属于肠道有益微生物,具有营养人体、增强机体免疫、改善胃肠功能等生理作用。Xu等[22]分析显示,在CHB和LC患者体内肠道双歧杆菌从有益菌转变为机会病原菌,与健康人群相比,CHB和LC患者肠道中双歧杆菌频率减少,LC中检出频率低于CHB患者。Bacteroidota和Firmicutes均为肠道主要菌群,HBV感染导致机体肠道细菌多样性下降,Bacteroidota下降,Firmicutes增多。Bacteroidota中含有胆汁酸盐水解酶,其可直接与胆盐和胆固醇等结合,从而降低机体胆固醇,故其下降可加重肝组织损伤,增加癌变风险[23]。Bacteroidota和Firmicutes相互作用下,肠道有效吸收食物中热量、积累脂肪,脂肪的积累会减少肠道黏蛋白,降低肠道屏障能力,增加Firmicutes侵袭风险,增加机体炎症反应,加重肝脏组织损伤和癌变风险。
综上所述,HBV感染者随着肝损伤病情加重可出现肠道菌群丰度和多样性下降,且HBV感染者肠道菌群丰度和多样性与AIH和PBC患者均存在明显差异。然而,受条件限制,本研究样本量纳入样本量偏小,导致研究结果存在一定局限性,需要后续加大样本量做进一步验证,以更好地做好CHB感染者疾病控制和不良结局预防。
利益冲突声明:所有作者均声明不存在利益冲突。
猜你喜欢
肝博士(2024年1期)2024-03-12 08:38:08
英语世界(2023年10期)2023-11-17 09:18:18
现代临床医学(2023年3期)2023-05-24 08:57:50
中老年保健(2022年2期)2022-08-24 03:20:50
科学(2020年4期)2020-11-26 08:27:06
科学大众(中学)(2019年3期)2019-05-17 10:04:30
汽车观察(2018年10期)2018-11-06 07:05:26
动物营养学报(2015年10期)2015-12-01 02:26:20
少儿科学周刊·少年版(2015年1期)2015-07-07 17:15:12
现代检验医学杂志(2015年4期)2015-02-06 02:02:11
