
lncRNA-TUG1靶向Zeste同源物增强子2对缺氧心肌细胞的影响*
2023-12-04 02:09常晨许玉莉任艳玲李金轶苏强
中国病理生理杂志 2023年11期
常晨, 许玉莉, 任艳玲, 李金轶, 苏强,2△
(1桂林医学院,广西 桂林 541000;2广西壮族自治区江滨医院,广西 南宁 530021)
心肌梗死(myocardial infarction, MI)是全球心血管疾病发病和死亡的主要原因。尽管心肌再灌注治疗已普遍开展,但冠状动脉血流恢复后不可避免的产生再灌注损伤,这不仅会加剧缺血部位损伤,还会牵连先前未受累的正常心肌组织[1]。氧化应激以及炎症反应被认为是MI诱导心肌细胞损伤的主要因素[2]。Zeste同源物增强子2(enhancer of Zeste homolog 2, EZH2)是一种组蛋白甲基移换酶,介导血管内皮细胞的形成[3]。此外,抑制EZH2可促进脑源性神经营养因子(brain-derived neurotrophic factor, BDNF)和内皮型一氧化氮合酶(endothelial nitric oxide synthase, eNOS)的表达[4]。研究显示过表达长链非编码RNA(long noncoding RNA, lncRNA)-牛磺酸上调基因1(taurine up-regulated gene 1, TUG1)可能是缺氧心肌细胞损伤的重要原因[5]。其中涉及的机制部分归因于eNOS和BDNF的蛋白表达水平降低,并可能进一步通过影响炎症反应和线粒体生物合成,参与心肌细胞损伤[6]。此外,我们前期研究[6]显示沉默TUG1有益于缓解MI后心功能损伤。然而,lncRNATUG1是否通过EZH2途径影响eNOS和BDNF的表达以及缓解缺氧心肌细胞中线粒体功能紊乱和炎症反应有待证实。因此,本研究构建HL-1细胞缺氧模型进行探究验证,进一步探索缺氧诱导的心肌细胞中lncRNA-TUG1对EZH2以及eNOS和BDNF的影响。此外,我们还探讨了沉默TUG1的表达对线粒体合成功能和炎症反应的影响,这为MI的治疗提供参考资料。
材料和方法
1 细胞系及试剂
HL-1心肌细胞系购自湖南丰晖生物科技有限公司。肌酸激酶同工酶(creatine kinase MB isoenzyme,CKMB)、心肌钙蛋白I(cardiac troponin I, cTnI)、肌红蛋白(myoglobin, MYO)、白细胞介素1β(interleukin-1β, IL-1β)、IL-6和肿瘤坏死因子α(tumor necrosis factor-α, TNF-α)ELISA试剂盒购自Cusabio;EZH2抗体(21800-1-AP)购自Proteintech;GAPDH抗体(ab22555)、histone H3抗体(ab1791)和兔IgG抗体(ab172730)购自Abcam;HRP标记的Ⅱ抗(A0216)购自Beyotime;SYBR®Premix Ex Taq、PrimeScriptTMRT试剂盒和总RNA提取试剂盒购自TaKaRa;高糖DMEM培养液、胎牛血清、青霉素-链霉素和胰蛋白酶购自HyClone;CCK-8试剂盒购自Beyotime;质粒中量抽提试剂盒购自杭州爱思进生物技术有限公司;ChIP试剂盒购自Millipore。所用引物由上海吉玛制药技术有限公司设计合成,序列见表1。
2 主要方法
2.1 细胞培养及分组处理 HL-1细胞用高糖DMEM培养液(含10%胎牛血清、1×105U/L青霉素和100 mg/L链霉素),并置于37 ℃、5% CO2恒温培养箱中培养。构建siTUG1和对照质粒siNC并分别转染至HL-1细胞内,随机分成常氧(normoxia)组、缺氧(hypoxia)组、hypoxia+siNC组和hypoxia+siTUG1组。将normoxia组置于常氧条件(37 ℃、5% CO2),其余组置于缺氧条件(1% O2、5% CO2、94% N2)下培养24 h[7]。
2.2 细胞转染实验 取对数生长期的细胞,调整密度至2×107/L,接种于96孔板中(每孔各100 μL)。细胞贴壁后转染siRNA。转染方法如下:(1)稀释质粒:用50 μL的Opti-MEM稀释200 μmol/L的siRNA,混匀后于室温孵育5 min;(2)稀释Lipo3000:用50 μL Opti-MEM稀释10 μL Lipo3000,混匀并室温孵育5 min。将(1)与(2)混匀并于室温孵育20 min,加入24孔板中(每孔各50 μL)后并置入培养箱培养6 h后,更换完全培养液培养。将培养皿放入培养箱中培养48 h后分装用于后续实验。
2.3 CCK-8实验 将对数生长期的HL-1细胞,以2×107/L密度接种于96孔板(每组6个复孔,每孔100 μL)。细胞贴壁后转染siNC和siTUG1。根据分组,将hypoxia组、hypoxia+siNC组和hypoxia+siTUG1组置于缺氧条件下培养。待24 h后,向培养液中加入20 μL的CCK-8溶液,培养箱中孵育2 h,酶标仪中450 nm处测定吸光度(A)值[8]。
2.4 ChIP实验 采用ChIP试剂盒,通过酶消化将细胞交联染色质超声破碎后于4 ℃、16 099.2×g离心10 min,除去不溶解的沉淀。将上清液转移至新的EP管中分为50 μL一份。将超声后的50 μL的样本置于冰上,加入450 μL稀释液(含2.25 μL蛋白酶抑制剂),混匀后移取5 μL上清液为内参照“Input”。以IgG为阴性对照。分别加入不同抗体和20 μL混匀的蛋白A磁珠后于4 ℃旋转孵育过夜。使用磁力架将蛋白A磁珠沉淀下来后洗涤并去除上清。进一步洗脱蛋白质-DNA复合物和反交联蛋白质-DNA复合物为单独的DNA,加入100 μL ChIP清洗缓冲液,62 ℃振荡孵育2 h后再于95 ℃孵育10 min[9]。然后将样品置于室温冷却,将提取的DNA纯化和RT-qPCR检测。
2.5 RT-qPCR实验 Trizol试剂盒提取总RNA后按照PrimeScriptTMRT试剂盒将RNA逆转录成cDNA。参照SYBR®Premix Ex Taq试剂盒检测PCR扩增产物,复孔检测后根据2-ΔΔCt法进行比较[6]。
2.6 Western blot实验 提取细胞蛋白,测定蛋白浓度。上样行电泳并湿转至PVDF膜。室温封闭结束后于4 ℃加入Ⅰ抗过夜孵育;次日用TBST洗膜后于室温加入Ⅱ抗孵育2 h,待再次洗膜后进行化学发光法显色,在化学发光成像仪下拍照分析。
2.7 ELISA实验 取各组细胞培养上清,按ELISA试剂盒说明书检测心肌损伤标志物cTnI、CKMB、MYO及炎症因子IL-6、IL-1β、TNF-α水平。96孔板中加入待测样本及标准品,于37 ℃恒温孵育,加入生物素标记的抗体孵育后加入HRP标记的抗生物素蛋白孵育。洗涤后加入TMB显色底物溶液于37 ℃避光显色,加入终止液终止反应,在450 nm波长处测定A值[6]。
3 统计学处理
每组实验至少重复3次,实验结果用均数±标准误(mean±SEM)表示。采用SPSS 24.0软件分析数据,GraphPad Prism 9.5作图。多组间比较采用单因素方差分析,两两比较采用LSD-t检验。以P<0.05为差异有统计学意义。
结果
1 lncRNA-TUG1在HL-1细胞系各分组中表达
如图1所示,缺氧可诱导HL-1心肌细胞lncRNA-TUG1表达显著升高(P<0.05)。此外,与hypoxia组和hypoxia+siNC组相比,hypoxia+siTUG1组lncRNA-TUG1表达显著降低(P<0.05)。

Figure 1. RT-qPCR measurement of the expression of lncRNATUG1 in the cells of different groups. Mean±SEM. n=3. **P<0.01 vs normoxia group; ##P<0.01 vs hypoxia+siNC group.图1 RT-qPCR检测lncRNA-TUG1在各组细胞中的表达
2 lncRNA-TUG1通过EZH2途径影响BDNF和eNOS的表达
ChIP分析显示,沉默TUG1的表达可进一步抑制EZH2与eNOS和BDNF启动子区域的结合(P<0.05),见图2。
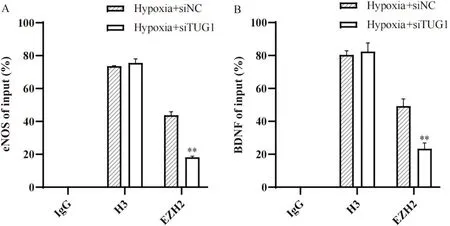
Figure 2. Effects of lncRNA-TUG1 on the interaction of EZH2 with the promoter regions of eNOS (A) and BDNF (B). IgG was used as a negative control and H3 as a positive control. Mean±SEM. n=3. **P<0.01 vs hypoxia+siNC group.图2 lncRNA-TUG1靶向EZH2与eNOS和BDNF启动子区域间的相互作用
3 沉默TUG1表达缓解缺氧对HL-1细胞活力的抑制
与normoxia组相比,hypoxia组和hypoxia+siNC组细胞活力明显降低(P<0.05);相较于hypoxia组和hypoxia+siNC组,hypoxia+siTUG1组细胞活力得到显著改善(P<0.05),见图3。

Figure 3. Evaluation of cardiomyocyte viability by CCK-8 assay.Mean±SEM. n=6. **P<0.01 vs normoxia group; ##P<0.01 vs hypoxia+siNC group.图3 CCK-8分析心肌细胞活力
4 沉默TUG1表达降低缺氧诱导的HL-1细胞心肌损伤标志物的表达水平
与normoxia组相比,hypoxia组和hypoxia+siNC组CKMB、cTnI和MYO均显著升高(P<0.05),而沉默TUG1表达能够部分逆转上述指标,见图4。

Figure 4. Measurement of markers of myocardial injury by ELISA. Mean±SEM. n=3. *P<0.05, **P<0.01 vs normoxia group; ##P<0.01 vs hypoxia+siNC group.图4 ELISA检测心肌损伤标志物
5 沉默TUG1表达促进缺氧诱导的HL-1细胞线粒体生物合成
与normoxia组相比,hypoxia组和hypoxia+siNC组线粒体生物合成相关转录调节蛋白核呼吸因子1(nuclear respiratory factor 1, NRF1)、过氧化物酶体增殖物激活受体γ辅激活因子1(peroxisome proliferator-activated receptor γ coactivator-1, PGC-1)和线粒体转录因子A(mitochondrial transcription factor A,TFAM)的表达水平显著降低(P<0.05);与hypoxia组和hypoxia+siNC组相比,hypoxia+siTUG1组NRF1、PGC-1和TFAM蛋白表达水平显著升高(P<0.05),见图5A。RT-qPCR结果同样显示,沉默TUG1后NRF1、PGC-1和TFAM的mRNA水平均显著回升(P<0.05),见图5B~D。
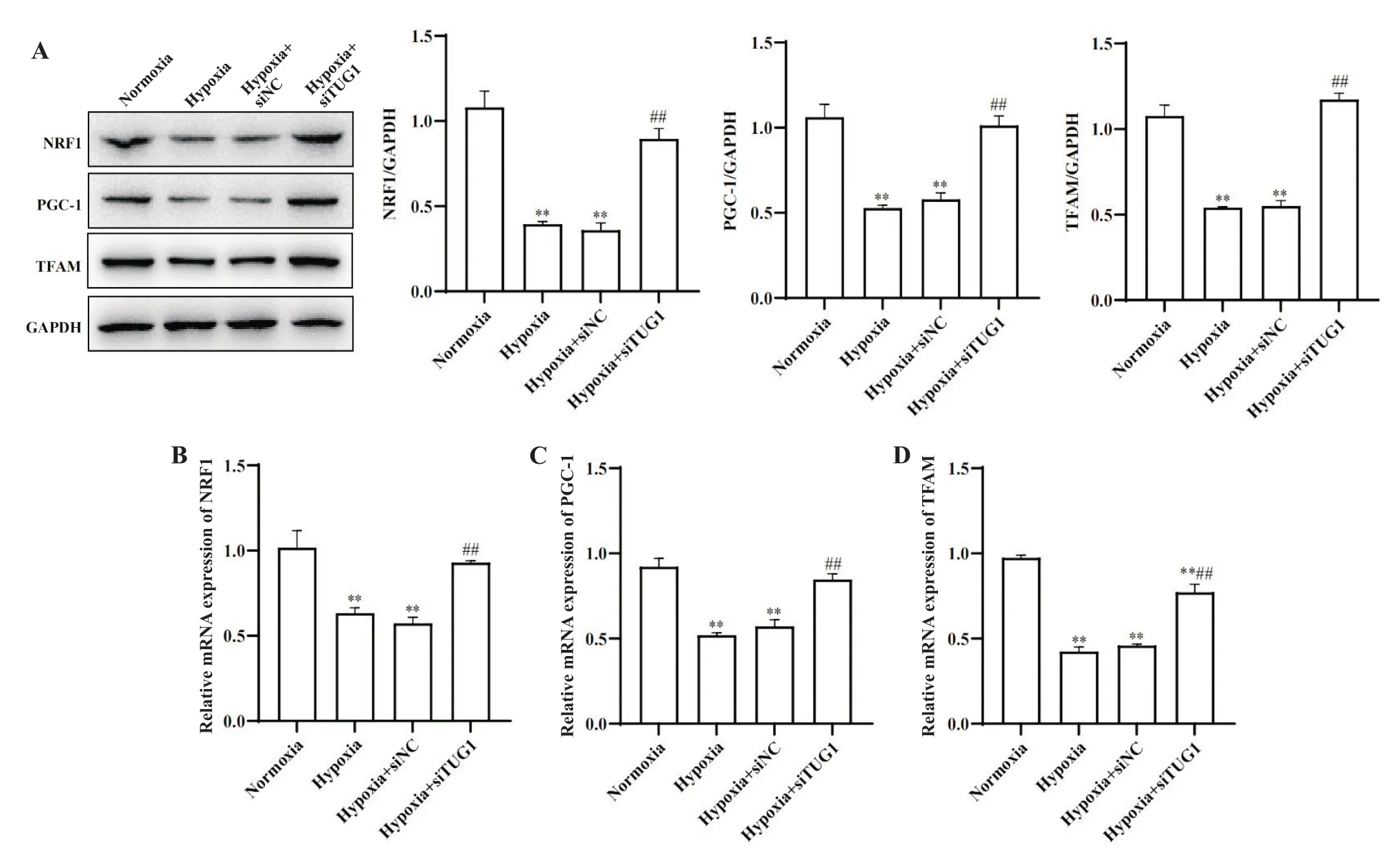
Figure 5. The expression of mitochondrial biosynthesis-related transcriptional regulator genes was detected by Western blot and RT-qPCR. A: Western blot showing the expression of mitochondrial biosynthesis-related proteins; B, C and D: the mRNA expression of mitochondrial biosynthesis-related genes. Mean±SEM. n=3. **P<0.01 vs normoxia group; ##P<0.01 vs hypoxia+siNC group.图5 Western blot和RT-qPCR检测线粒体生物合成相关转录调节基因
6 沉默TUG1表达抑制缺氧诱导的HL-1细胞炎症反应
RT-qPCR结果显示,与normoxia组相比,hypoxia组和hypoxia+siNC组IL-1β、IL-6和TNF-α的mRNA表达显著升高(P<0.05);与hypoxia组和hypoxia+siNC组相比,hypoxia+siTUG1组IL-1β、IL-6和TNF-α的mRNA表达受到显著抑制(P<0.05),见图6A~C。ELISA结果显示,沉默TUG1能显著抑制炎症因子IL-1β、IL-6和TNF-α的释放(P<0.05),见图6D~F。
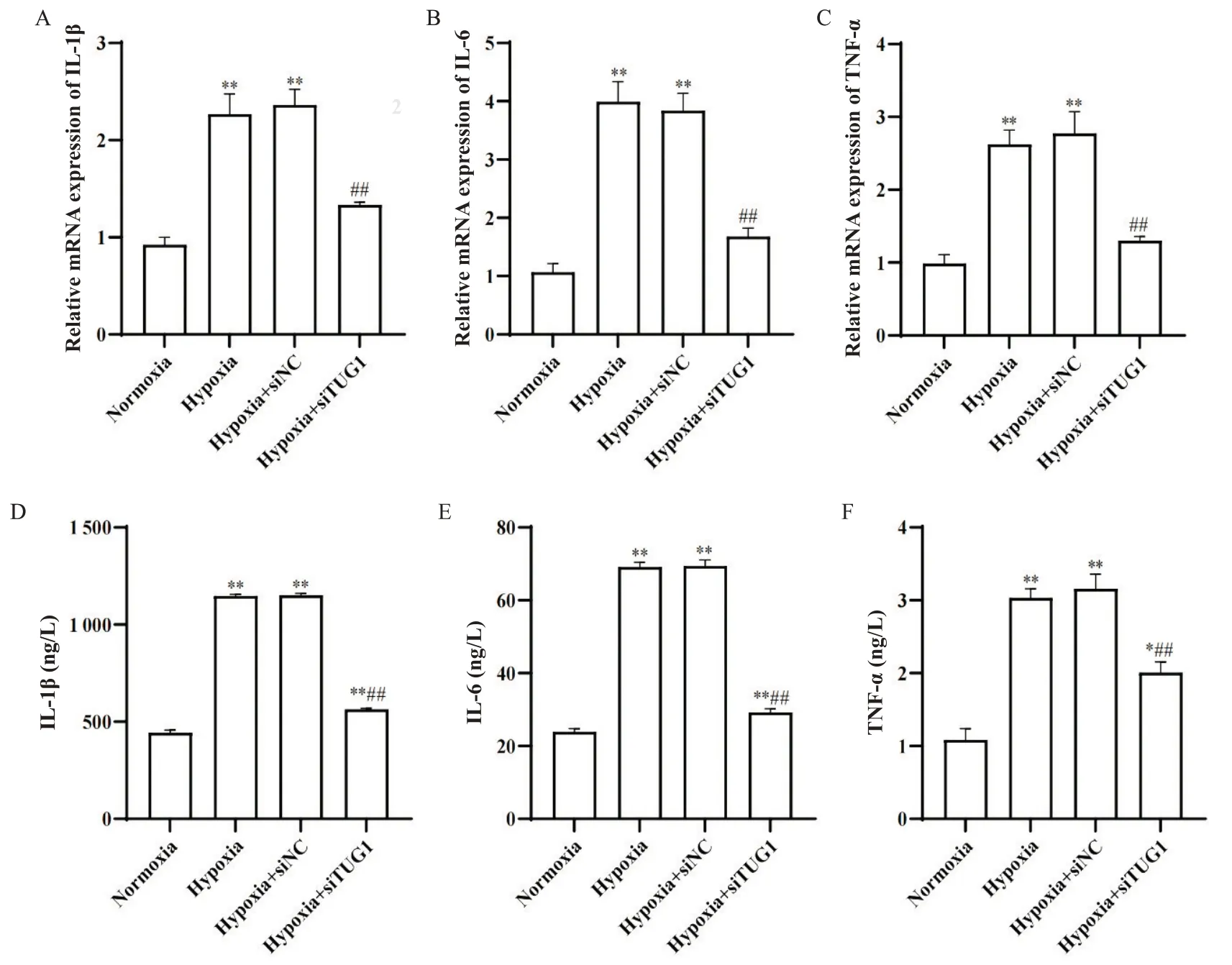
Figure 6. RT-qPCR and ELISA for inflammatory factors. RT-qPCR showing the mRNA expression of inflammatory factors: IL-1β(A), IL-6 (B) and TNF-α (C); ELISA for inflammatory factors: IL-1β (D), IL-6 (E) and TNF-α (F). Mean±SEM. n=3. *P<0.05, **P<0.01 vs normoxia group; ##P<0.01 vs hypoxia+siNC group.图6 RT-qPCR和ELISA检测炎症因子
讨论
TUG1位于人染色体22q12.2,其转录产物是长度为7.1 kb的lncRNA[10]。lncRNA-TUG1被认为是心血管疾病预后不良的生物标志物[11]。lncRNA-TUG1在缺氧心肌细胞中过表达,并可能通过刺激ROS的产生,加重心肌细胞损伤[12]。此外,先前研究[6]显示在小鼠MI模型中沉默TUG1可进一步通过抗炎发挥心肌保护作用。部分可能归因于沉默TUG1表达后进一步上调了eNOS和BDNF的水平[6]。然而,其中具体机制尚不明确。本研究构建HL-1细胞缺氧模型,通过沉默TUG1表达,进一步探索缺氧诱导的心肌细胞中lncRNA-TUG1对EZH2以及eNOS和BDNF的作用。此外,我们还探讨了沉默TUG1的表达对缺氧心肌细胞的线粒体合成功能和炎症反应的影响。
本研究结果显示沉默TUG1的表达部分逆转了缺氧对心肌细胞活力的抑制。已有研究指出过表达lncRNA-TUG1可能靶向EZH2参与多种肿瘤的发生与转移[13]。此外,Mitić等[14]在小鼠肢体缺血模型中观察到抑制EZH2的表达后可能进一步促进了eNOS和BDNF的表达。本研究结果显示沉默TUG1表达可能抑制EZH2与eNOS、BDNF启动子区域的结合。EZH2被认为是巨噬细胞激活和自身免疫性炎症的重要调节因子[15]。eNOS是调节心血管稳态的重要调节因子,介导血管内皮NO的合成[16]。缺血可诱导血管内皮细胞中NO合成限速酶eNOS的激活,促进NO生成发挥舒张血管的作用[17]。此外,在脓毒性心肌病的小鼠模型中观察到BDNF可通过刺激eNOS进而抑制氧化应激和细胞凋亡发挥心肌保护作用[18]。我们先前研究[6]显示沉默TUG1后,小鼠心肌组织中eNOS和BDNF的表达上调。此外,部分研究[19-20]表明eNOS和BDNF表达水平的增高可以降低动脉粥样硬化模型中炎症因子的表达。本研究结果显示沉默TUG1表达可抑制EZH2与BDNF和eNOS启动子区域的结合,这可能是沉默TUG1降低缺氧心肌细胞炎症的潜在机制。心肌细胞损伤心脏维持正常功能所需的ATP超过95%源自线粒体的氧化磷酸化[21]。PGC-1α可通过NRF1和TFAM来诱导线粒体生物发生[22]。沉默TUG1可进一步上调miR-26a缓解LPS诱导的线粒体损伤及炎症反应[23]。同样,本研究结果显示沉默TUG1可进一步促进缺氧心肌细胞线粒体生物合成相关转录调节蛋白NRF1、PGC-1和TFAM的表达。这说明沉默TUG1表达对缺氧诱导的HL-1细胞线粒体生物合成具有保护作用。
综上所述,lncRNA-TUG1可能通过EZH2途径参与线粒体功能紊乱和炎症反应的过程。当lncRNATUG1表达降低时,其与EZH2的结合减弱,进而影响EZH2与eNOS和BDNF启动子区的结合。因此,沉默TUG1可能通过EZH2途径影响eNOS和BDNF的表达并缓解缺氧诱导的HL-1细胞炎症反应。此外,沉默TUG1可上调线粒体合成相关蛋白NRF1、PGC-1和TFAM的表达,促进线粒体生物合成。总之,沉默TUG1可能通过EZH2途径减轻缺氧心肌细胞的线粒体功能紊乱和炎症反应。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
海南医学(2016年8期)2016-06-08
中国病理生理杂志(2015年8期)2015-12-21
华南农业大学学报(2015年5期)2015-12-04
癌变·畸变·突变(2014年1期)2014-03-01
