
以生物标志物为导向的临床试验研究*
2023-11-30 01:24宋佳丽孙凤宇刘芝霖
中国卫生统计 2023年5期
宋佳丽 孙凤宇 刘芝霖 李 康 侯 艳△
近年随着精准医疗的发展,越来越多的药物针对疾病的分子作用机制和特定靶点进行开发。在创新药物研发上,以生物标志物为导向的临床试验研究,即探索与药物疗效或安全性相关的生物标志物,根据其识别亚组并制定个体化治疗的临床试验方案,从而使患者接受更有益的治疗、加速药物研发进程、最大限度地规避新药研发的风险,已引起人们的极大关注[1-3]。本文将对以生物标志物为导向的临床试验设计方法和特点进行简要综述,以期对创新药物研究起到一定的助力作用。
生物标志物在临床试验中的作用
生物标志物(biomarker)是指在正常生理、病理过程中的某些指标,或对暴露或治疗干预措施产生的某种应答或反应,其通常能够进行客观测量和评价,主要包括分子、组织学、放射学或生理学等特征[4]。2016年,美国食品药品监督管理局(food and drug administration, FDA)和美国国立卫生研究院发布了生物标志物、结局指标和其他工具(biomarkers, endpoints, and other tools, BEST)术语表等[5]。其中根据功能定义了七种生物标志物,即诊断、监测、药效学/反应、预测型、预后型、安全性、易感性/风险生物标志物。中国国家药品监督管理局药品审评中心2021年6月发布了《生物标志物在抗肿瘤药物临床研发中应用的技术指导原则(征求意见稿)》[6],其中定义了六种生物标志物。相比之下,美国FDA多了易感性/风险生物标志物。一个生物标志物在不同的应用场景下,可能具有不同的功能,从而会归属于不同的分类。以生物标志物为导向的临床试验研究最常使用的是预测型和预后型生物标志物,前者主要指能够鉴别对研究药物最可能有应答反应的生物标志物,后者则是指更有可能观察到终点事件或疾病进展的高风险人群的生物标志物。
以生物标志物为导向的临床试验研究的关键是,通过合适的生物标志物确定亚组人群。此时,相关标志物检测方法的可靠性十分重要。理想情况下,试验中生物标志物的检测方法,应该对高风险或药物敏感者(目标患者)有较高的灵敏度,同时对低风险或对药物无应答者(非目标患者)有较高的特异度。如果检测方法的灵敏度差,则目标患者被筛选入组的可能性降低,从而延缓整个研究入组的进度;如果检测方法特异度较低,则选入的非目标患者可能较多,从而稀释了药物的治疗效果。针对生物标志物的检测方法可以研发出伴随诊断产品(companion diagnostic, CDx)。目前已有FDA批准的CDx用于指导精准用药[7]。CDx通常是一种体外医疗设备,可为专业人员提供相关信息,保障安全、有效地使用相应的药品或生物产品,常用于确定:(1)最有可能从治疗中受益的患者;(2)可能因接受治疗而导致严重不良反应风险增加的患者;(3)对治疗安全且有效的患者。目前,CDx可以与药物研发同步进行,或在药物上市后进行开发。
临床试验生物标志物的识别与确证
生物标志物的识别可以通过回顾性或前瞻性研究获得相关信息,并运用多种统计学或机器学习方法进行分析。基于既往研究数据识别生物标志物是一种回顾性研究,即主要使用已经完成的各种临床试验数据,包括随机对照试验(randomized controlled trial,RCT)或队列研究数据进行一个或一组生物标志物的识别。通过回顾性分析,找到相关的生物标志物及其阳性患者亚组治疗有效的证据,这在一定程度上避免了伦理学上的问题。此外,也可利用失败的临床试验中采集到的血液或组织学样本,结合最新研究进展,进行新的生物标志物检测及分析探索,随后通过富集设计进一步研究,有可能使药物因亚组人群获益而批准上市。例如,一项转移性结直肠癌患者的随机Ⅲ期临床试验中,预测型生物标志物KRAS可用于预测帕尼单抗(panitumumab)和西妥昔单抗(cetuximab)的疗效。对该项目进行回顾性分析发现,92%患者可评估KRAS状态,其中43%具有KRAS突变。针对无进展生存期指标,在野生型和突变亚组人群中,帕尼单抗治疗相较于最佳支持治疗的风险比(hazard ratio, HR)分别为0.45(95%CI: 0.34~0.59)和0.99(95%CI: 0.73~1.36)[8],为KRAS野生型亚组的治疗有效性提供一定的证据支持。值得注意的是,如果使用来自两个或以上独立RCT数据进行回顾性分析,其获得的结果证据力度更强。生物标志物的前瞻性研究主要是根据早期研究中确定的、具有已知病理生理效应的蛋白质或遗传标志物,或者根据Ⅰ/Ⅱ期临床试验中标志物分析结果等,进一步通过确定阈值等方法,预先进行设计,对已经识别出的潜在生物标志物进行探索或验证。
以生物标志物为导向的试验设计
使用前瞻性试验验证药品或医疗器械有效性和安全性的力度远大于回顾性研究。目前已经开发并使用了多种以生物标志物为导向的前瞻性临床试验策略及设计方法,主要有简单富集设计(classical enrichment design)、纳入所有受试者设计(all-comers design)、适应性富集设计(adaptive enrichment design)以及主方案设计(master protocol design)等。
1.简单富集设计
即仅对最有可能从治疗中受益且具有特定分子特征的患者进行研究,其主要思想是在临床试验中根据受试者的生物学特征,前瞻性地识别或定义从研究药物中获益最大的目标人群,从而提高临床试验效率的一种策略和设计方法(图1)。比如抗肿瘤治疗药物通常针对癌细胞表面或细胞内相关的受体、酶、激素或其他内源活性物质,对此可将一个或多个相应的生物标志物作为选择富集人群的依据。例如,对于人表皮生长因子受体2(HER2)蛋白阳性的乳腺癌手术切除患者,使用曲妥珠单抗(trastuzumab)联合紫杉醇(paclitaxel)治疗,能够显著提高其无病生存期[9]。一般而言,仅以标志物阳性受试者为目标人群,需要充分说明该亚群患者可以从研究药物中获益,而生物标志物阴性的患者不会从该治疗中受益,即药物对标志物阴性患者无效或存在安全性问题[10]。常见的设计主要包括:(1)通过预后型生物标志物,识别更有可能或更快观察到终点事件的高风险人群;(2)根据预测型生物标志物,选择最有可能对研究药物应答或对现有对照药物无应答的受试者;(3)同时使用预后型和预测型标志物以减少受试者和试验结局的异质性。
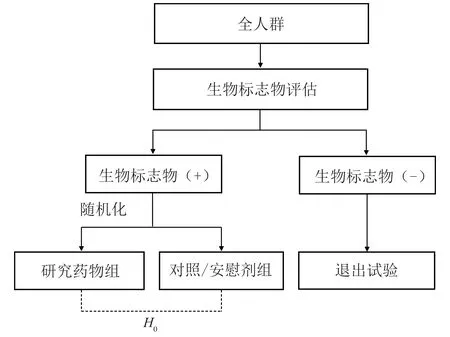
图1 简单富集设计示意图
2.纳入所有受试者设计
如果药物的作用机制或已有数据表明,生物标志物阳性患者较阴性患者的疗效更好,在治疗毒性较小的情况下,可同时纳入生物标志物阳性和阴性的患者。这种策略的优点是,能在非富集人群中进行合理的获益-风险评估,避免了由于标志物阳性人群获益较高而得出全人群获益的结论,贻误原本没有或较小获益的标志物阴性人群的治疗[11],主要包括:
(1)顺序检验策略设计(sequential testing strategy design):这种设计将标志物阳性和阴性患者同时纳入试验,但是需要预先设定研究假设。其主要包括两种检验策略:(1)先在全人群中检验研究药物和对照组是否有差异(H0(2)),如果在预设的检验水准α1下,差异没有统计学意义,则使用初始设定的检验水准α2,在预定义的标志物阳性亚组中进行检验(H0(1));(2)先在标志物阳性亚组中进行检验(H0(1)),若差异有统计学意义,进一步在整个人群中进行检验(H0(2))(图2),同时应该注意控制总Ⅰ类错误概率[12]。当假设治疗对整体有效,亚组分析仅作为补充分析时,一般推荐使用前者;当有较强的证据支持在生物标志物阳性亚组中的治疗效果更好,且能够入组足够的标志物阳性人群时,建议使用后者。检验的具体方法可有Bonferroni单步检验法、逐步多重检验法(stepwise multiple procedure)和回退法(fallback procedure)等。

图2 顺序检验策略设计示意图
(2)生物标志物分层设计(biomarker-stratified design):即根据生物标志物状态分层随机化,将患者分配到不同的干预组中(图3),根据标志物分层随机化可以确保参与研究的不同生物标志物患者都能接受到治疗,且能够更好地保证组间均衡[13]。该设计预先定义一个或一组目标生物标志物,并对具有生物标志物检测结果的患者进行前瞻性分层随机化。其更适用于有候选生物标志物,但前期没有充分证据支持只纳入标志物阳性人群的情况。一个典型的案例是厄洛替尼(erlotinib)在非小细胞肺癌(NSCLC)中的生物标志物验证试验(MARVEL)。之前研究发现表皮生长因子受体(EGFR)可能是治疗NSCLC的重要生物标志物[14],故该试验根据EGFR免疫组化测量结果将二线治疗患者随机分配至培美曲塞(pemetrexed)或厄洛替尼治疗组。本设计同样有两个研究假设,即针对全人群和标志物阳性亚组人群,可以使用前述的任一种多重检验方法进行检验。

图3 生物标志物分层设计示意图
(3)混合设计(hybrid design):若开展试验前有充分且有力的证据表明,试验治疗对生物标志物阴性亚组有不利影响,或在该亚组中可能存在过度毒性时,将这一类亚群患者随机分配接受不同组别治疗,或使其退出试验显然不符合伦理,此时可以考虑使用混合设计[15](图4)。该设计在生物标志物阳性亚群中进行随机化,能够检验研究药物在该亚组中的效果,类似于生物标志物分层设计。然而,与其不同的是,生物标志物阴性亚群均接受标准治疗。此外,当事先存在可信的证据证明相关生物标志物阳性亚组存在疗效时,也适合选择这种设计,对具有某特定标志物的患者随机分配治疗。混合设计主分析主要针对生物标志物阳性亚组,而生物标志物阴性亚组仅作为辅助分析使用。
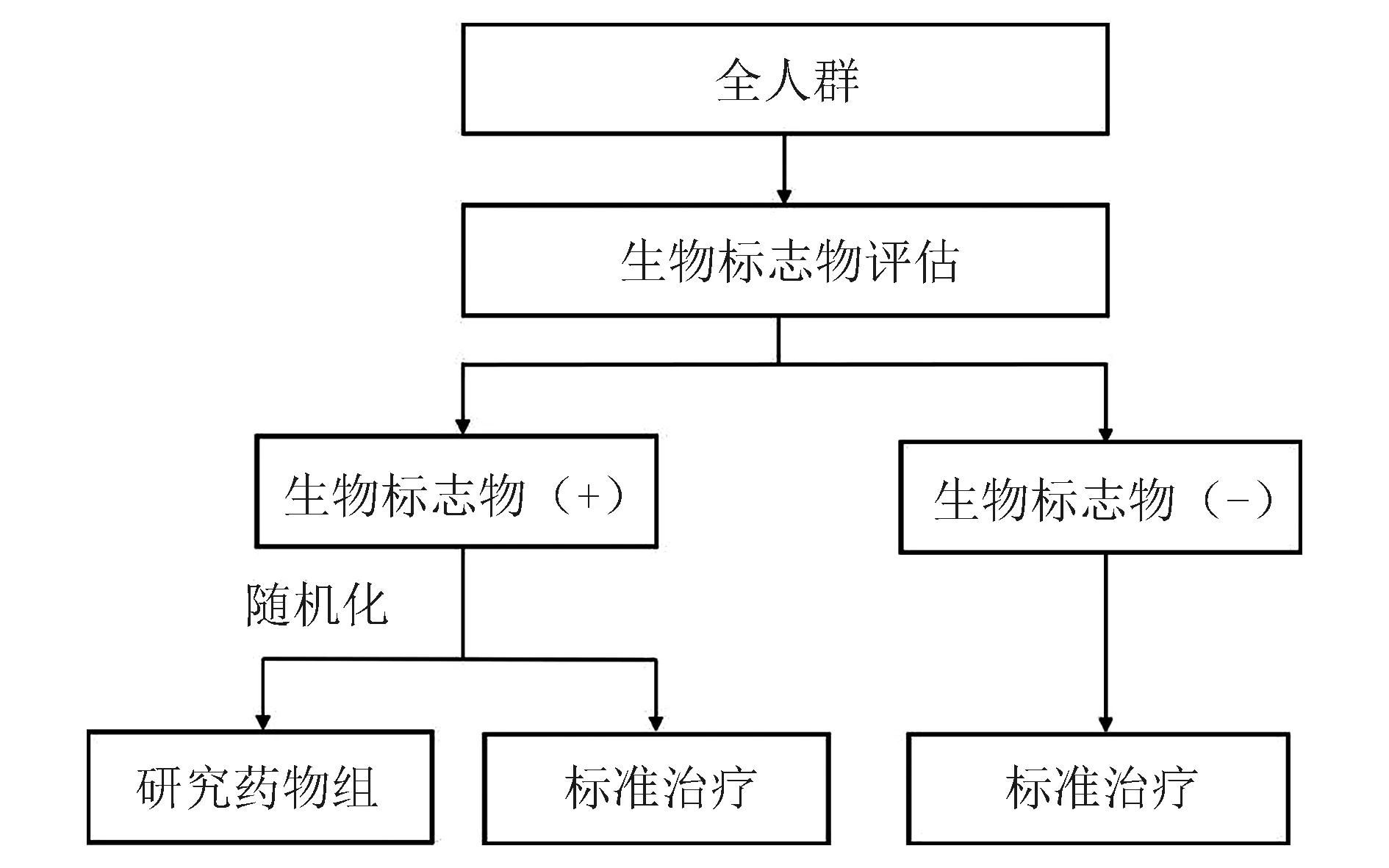
图4 标志物阳性和阴性受试者皆获益的混合设计
3.适应性富集设计:若在设计阶段对于靶向生物标志物了解不充分,如研究药物在生物标志物阳性患者人群中的疗效、阳性患者在目标人群中的比例等具有较大的不确定性,则可以使用一种更为灵活的试验设计方法,即适应性富集设计[16]。其前瞻性地规定如何根据期中分析结果对后续实践过程进行修改,包括减少或限制标志物阴性亚组患者入组等[17],方便在试验后期调整相应内容(图5)。常见的设计类型有:

图5 适应性富集设计示意图
(1)适应性调整患者入组条件(adaptive patient enrollment modification):一项随机对照试验可以同时入组生物标志物阳性和阴性的患者,在主要终点是标志物阳性亚组的疗效时,若期中分析结果显示标志物阴性患者人群治疗效果远低于标志物阳性患者的疗效,则可减少或完全停止标志物阴性的患者入组[18]。然而,对于早期探索性研究,最好同时纳入该预测型生物标志物阴性的患者,从而评估药物在整体患者人群中的获益与风险。当无法确定一个生物标志物是否具有预测作用时,主要终点可以是全人群中的效应。
(2)适应性阈值策略(adaptive threshold strategy):假设在试验开始时有一个预先指定的生物标志物检测,期望找到一个阈值,将连续变量转换成二分类分组变量,使得按这个分组变量划分的两个组别的疗效差异最大[19]。由于定义亚组的界值可能并不确定,可以首先比较研究药物组和对照组所有患者疗效,如果在预先指定的检验水准α1下得出两组差异显著的结论,则认为研究药物对所纳入的患者整体有效。否则,使用预先确定的检验水准α2进行第二阶段检验,寻找最佳判定阈值,使生物标志物阳性患者和阴性患者间的疗效差异最大化。
(3)适应性特征设计(adaptive signature design):此设计常在方案中事先制定研究策略与Ⅰ类错误控制方法等,并进行一系列的统计建模与分析,获得关于标志物的预测特征。例如,使用多个生物标志物建立预测药物敏感性的分类器,在期中分析时分析预先规定的终点,确定最佳的诊断阈值。该方法需要将数据分为训练集和测试集,使用训练集定义最有可能从研究药物治疗中获益的患者特征,然后对测试集中具有该特征的患者的疗效进行评估[20]。
(4)适应性无缝设计(adaptive seamless design):即将几个原本独立的试验组合成一项研究的试验设计,该设计使用全部数据进行最终分析,可同时满足几个试验目的[21]。常见的两阶段适应性无缝设计通常包括学习阶段和确证阶段。学习阶段可对生物标志物进行探索,并根据累积的数据在本阶段结束时调整后续试验设置;确证阶段着重对上一阶段发现的生物标志物亚群早期疗效进行验证[22]。相较于传统方法,本设计可以减少各研究阶段的间隔时间,并充分利用从试验中收集到的所有数据评估药物疗效。
4. 主方案设计
主方案设计是指在一个临床试验中,同时对多个患者群(如不同的肿瘤)进行评估,或同时研究多种药物对一种疾病的疗效,需要预先设计一个具有多个目标的总体方案[23],其主要包括以下几种设计。
(1)篮式试验设计(basket trial design):该设计针对具有相同生物标志物特征的多种疾病或组织学类型患者,可视为多个单病种富集试验的组合,目前常用于抗肿瘤药物研究[24]。篮式设计仅纳入生物标志物阳性患者,并对其进行随机化分组或使用单臂研究,目前常用的是Ⅱ期单臂设计。然而,单臂研究一般要求主要终点是客观指标,因为在没有对照组时,通常认为客观指标的可解释性更强。例如,使用篮式设计对威罗非尼(vemurafenib)治疗BRAFV600突变的多种非黑色素瘤癌症疗效和安全性进行探索,其主要终点为影像学的客观缓解率[25]。
需要注意的是,在具有相同生物标志物的不同疾病或组织学类型患者间,研究药物可能存在一定的疗效差异。例如使用威罗非尼治疗BRAFV600突变患者时发现,当原发部位是黑色素瘤时治疗反应很高,而当患者原发部位是结直肠癌时,治疗反应却很低[25]。因此,对于篮式试验设计的研究,主分析一般是对不同的疾病或组织类型单独分析,然后在此基础上再进行综合分析。
(2)伞式试验设计(umbrella trial design):主要针对单一疾病或组织学类型患者进行多种治疗的有效性和安全性评估。该设计需要预先制定并评估一个明确的规则,通过规则匹配生物标志物人群和候选治疗方案。在检测患者生物标志物后,根据生物标志物种类和规则将患者分配到其对应的特异性部分试验中,各部分试验可以使用单臂或随机分组设计。一个典型例子是整合生物标志物的肺癌靶向治疗方法评价(BATTLE),该试验使用伞式设计在进展性NSCLC患者人群中,以四种生物标志物为导向,研究三种单抗和一个联合疗法治疗效果[26]。
(3)平台试验设计(platform trial design):融合了篮式试验和伞式试验的特点,可针对多种疾病或组织学类型评估多种临床研究药物或药物联合方案,从而找到某种疾病的最佳治疗方法[27]。这种设计方案具有很大的灵活性,有助于停止无效或不良反应发生率高的试验、尽快推进有利的早期结果至Ⅱ/Ⅲ期研究、引入新的部分试验研究等。在一项基于影像学和分子生物标志物确定局部晚期乳腺癌的新辅助治疗方案的研究(I-SPY 2)中[28],利用平台设计在三种生物标志物定义下的八个早期高风险亚组人群中,探索五种药物的治疗效果。
基于生物标志物临床研究的一般考虑
1. 生物标志物的证据强度
当预期使用以生物标志物为导向的临床试验设计时,需要对潜在预测/预后型生物标志物的临床前证据强度进行评估,以支持试验设计方法的选择。如果有较强的证据表明,试验治疗不能为所有患者提供益处,且仅限于生物标志物阳性患者获益,即治疗在生物标志物阳性人群中的应答率更高,或在生物标志物阴性患者中具有较大的毒性,则可以采用富集策略。否则,更适合纳入所有受试者的策略,避免错过可能获益的受试者,使药物具有更大的获益人群。
2. 生物标志物阳性率及其检测方法
在进行以生物标志物为导向的临床试验设计时,首先应该充分考虑生物标志物阳性人群在全人群中所占比例。如果比例过低,使用富集设计容易造成临床试验受试者招募困难,延长试验时间,不利于临床实施与执行,也会增加资源消耗,需要谨慎决策;如果比例过高,可能不需要进行富集试验设计,此时传统的临床试验方案更简单易行,且有更大的适应症人群。其次,为了准确识别出生物标志物阳性和阴性人群,应选择较高灵敏度和特异度的检测方法,且该技术应具有较好的有效性和重现性。最好在试验设计过程中,考虑对伴随诊断产品进行同步开发和验证。
3. 临床试验设计与执行程度
以生物标志物为导向的临床试验设计均具有较高的灵活性,因此能够锁定获益人群,加速药物开发速度。但是,如果在试验设计时对各种可能的情况及其相关解决措施考虑不周,或在试验实施过程规范化管理与质控上做得不够好等,极有可能导致试验数据质量差,用其分析得到的结论与事实相反,反而不利于患者受益。因此,需要建立相关规范文件,明确在临床试验各个阶段应充分考虑的问题。
4. 总Ⅰ类错误概率控制
以生物标志物为导向的临床试验研究在统计检验时,可能出现Ⅰ类错误概率膨胀问题,即由于在全人群和亚组人群中进行多次检验,使得治疗虽在研究人群中没有效果,却错误地得出疗效显著的统计结论。因此,必须使用合适的方法对其进行控制。当有充分证据表明生物标志物阳性亚组的疗效优于全人群疗效时,可以考虑使用固定检验顺序的方法(如守门法),即在α水平检验亚群疗效,若有显著意义则仍在α水平检验全人群疗效,否则停止检验[29];在没有足够的数据用来估计亚组和全人群的疗效时,可以考虑使用逐步多重检验法(如Hochberg和Hommel法)[30]或回退法[31]。
5. 知情同意书可理解性
相较于传统设计,以生物标志物为导向的临床试验设计常较为复杂、不容易解释,导致此类知情同意书篇幅通常较长,往往无法保证所有入组患者完全理解知情同意书中的相关内容,需加强患者的知情同意教育[32-33]。为此,可在早期让患者或试验中心参与知情同意书的审阅,使其尽量简化以便入组受试者理解;当设计有多个阶段或部分试验时,可让患者分别签署两个知情同意书,其中一个主要阐明治疗阶段或入组的情况,另一个则主要告知患者治疗可能获得的受益与风险。
展 望
目前,加快从实验室到临床应用的药物研发是临床研究发展的必然趋势。基于前期药理和毒理学等探索,充分认识患者的异质性,并通过伴随诊断产品辅助以生物标志物为导向的临床设计实施,可提高试验效率,对于加速药物研发起着关键作用。这种研究还可以在临床试验过程中寻找新的替代终点,缩短临床试验周期;或辅助研究者找到药物的敏感人群与获益人群,使更多人更早获益,并加速药物上市;同时,可以识别对药物治疗无效或安全风险较高的患者,减少其不必要用药与损伤。然而,由于生物系统的复杂性,如何准确找到相应的生物标志物,仍面临巨大的挑战。相信随着以生物标志物为导向的临床试验实施增加,会不断批准更多的创新药物。
猜你喜欢
动物营养学报(2024年2期)2024-03-05
实用心脑肺血管病杂志(2023年12期)2023-12-14
园林科技(2021年3期)2022-01-19
基层中医药(2020年5期)2020-09-11
心电与循环(2020年1期)2020-02-27
中华老年多器官疾病杂志(2016年9期)2016-04-28
医学研究杂志(2015年7期)2015-06-22
现代检验医学杂志(2015年6期)2015-02-06
现代检验医学杂志(2015年5期)2015-02-06
中国合理用药探索(2012年2期)2012-03-20
