
PHIP基因突变所致Chung-Jansen综合征一例
2023-11-28 19:04王慧超李田华杨柳卢园园
新医学 2023年11期
关键词:儿童
王慧超?李田华?杨柳?卢园园
【摘要】Chung-Jansen综合征(CHUJANS)是一种常染色体显性遗传病,是新近发现的罕见肥胖综合征,主要表现为发育迟缓、智力障碍、肥胖和畸形。该文报道1例以肥胖、睾丸小为主要表现的CHUJANS患儿,该患儿发育迟缓、智力障碍,伴有左肾缺如及低促性腺激素性性功能减退,基因检测结果提示PHIP基因突变,突变位点c.600+1G>C,最终诊断为CHUJANS。经过长期综合性治疗,患儿远期生活质量获得极大改善。CHUJANS发病率低,且累及多系统,该例扩展了CHUJANS的基因突变谱,有助于提高临床医师对该疾病的认识水平,及早识别并给予干预将有助于改善患者预后。
【关键词】Chung-Jansen综合征;PHIP基因;杂合突变;儿童
A case of Chung-Jansen syndrome caused by PHIP gene mutation Wang Huichao△, Li Tianhua, Yang Liu, Lu Yuanyuan. △970 Hospital of the PLA Joint Logistic Support Force, Weihai 264299, China
Corresponding author, Lu Yuanyuan, E-mail: yuan_yuan26@163.com
【Abstract】Chung-Jansen syndrome (CHUJANS), an autosomal dominant genetic disorder, is a newly discovered rare obesity syndrome, mainly manifesting as developmental delay, mental retardation, obesity and dysmorphism. We reported one CHUJANS child with obesity and small testes as the main manifestations. The patient had developmental delay, mental retardation, complicated with left renal agenesis and hypogonadotropic hypogonadism. Genetic testing prompted PHIP gene mutation at c.600+1G>C. The child was diagnosed with CHUJANS. After long-term comprehensive treatment, the long-term quality of life was significantly improved. As Chung-Jansen syndrome is low in prevalence and multi-systemic, this case report expands the spectrum of mutations in CHUJANS, which can deepen clinicians understanding of this disease. Early diagnosis and intervention contribute to enhancing clinical prognosis.
【Key words】Chung-Jansen syndrome; PHIP gene; Heterozygous mutation; Children
Chung-Jansen綜合征(CHUJANS,OMIM#617991)是一种以发育迟缓、智力障碍、肥胖和畸形为特征的常染色体显性遗传病,由位于6q14染色体上的PHIP基因(OMIM#612870)中的杂合突变所致,可在婴儿期发病,大多为基因突变从头合成,少数为家族遗传[1]。CHUJANS罕见,2012年de Ligt等[2]对100例智力障碍患者进行外显子测序,发现了1例PHIP基因突变所致的智力障碍患者,随后国外相继报道相关病例,现全球约有300例CHUJANS患者,但笔者见目前国内仅有1例报道[3]。本文报道了1例PHIP基因突变所致的CHUJANS男性患儿,除特征性表现外,该患儿还同时具有低促性腺激素性性功能减退症及左肾缺如。现总结该例患儿的临床特点及遗传学资料,并对相关文献进行复习总结,以提高临床医师对该病的认识,避免误诊漏诊。
病例资料
一、一般资料
患儿男,12岁5个月,汉族,非近亲婚配独生子,以肥胖、睾丸小为主要表现于2021年10月来中国人民解放军联勤保障部队第九七〇医院就诊。患儿通过剖宫产娩出,出生时体质量>5 000 g,在婴儿期无喂养问题,无长期服用药物史,患儿发育落后,12月龄说话,1岁6个月可走,学习成绩差,平日少言,与人沟通交际能力差,活动量少,易疲劳,家属未在意,未予特殊处理。近期家属无意间发现患儿睾丸小,遂到当地医院就诊,睾丸彩色多普勒超声检查(彩超)提示双隐睾,肾脏彩超显示患儿左肾缺如。家属诉患儿存在嗅觉异常。患儿既往史、家族史无特殊。
二、体格检查
体型肥胖,体质量78.4 kg(>P97),身高150.2 cm
(P10~P25),BMI 34.8 kg/m2(>P97),皮肤粗糙,耳垂厚,上眦赘皮,杏仁眼,下巴略尖、稍前凸,面部可见散在黑色素痣。双侧乳房隆起,未触及明显硬核。阴毛、腋毛稀疏,睾丸体积0.5~1 mL,外阴PH1期,阴茎长4.0 cm,周长5.5 cm。
三、诊疗经过
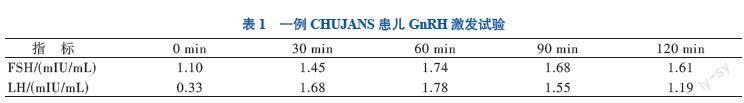
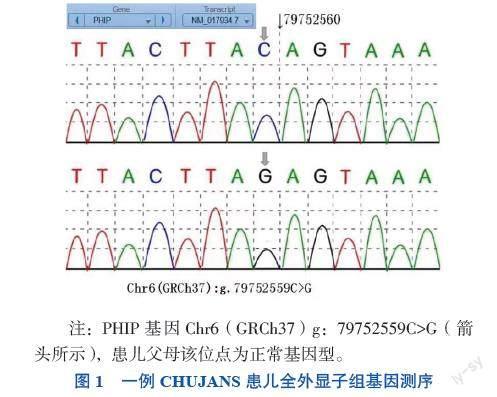
入院后测卵泡刺激素1.10 mIU/mL,黄体生成素0.33 mIU/mL,睾酮0.19 ng/mL,促性腺激素释放激素(GnRH)激发试验提示性腺轴尚未启动,见表1。复查睾丸彩超提示双隐睾,睾丸位于双侧腹股沟区,左侧大小约17 mm×9 mm,右侧大小约18 mm×8 mm,随呼吸上下游走,可部分进入阴囊。嗅觉测试存在轻度嗅 觉障碍,初步诊断为低促性腺激素性性功能减退症,给予肌内注射人绒毛膜促性腺激素(HCG)治疗。双侧乳腺彩超提示乳核可见,右侧大小约7 mm×4 mm,左侧大小约6 mm×5 mm。颅脑MRI未见异常。综上,患儿表现为异常的体质量增加、生长发育迟缓、低促性腺激素性性功能减退症、双隐睾及左肾缺如,高度怀疑患儿存在基因缺陷疾病,在取得患儿父母同意的情况下,对患儿及其父母抽血进行全外显子组基因测序。结果显示,患儿PHIP基因有1个杂合突变,该突变位点位于6号染色体PHIP基因内含子区域的一处杂合位点变异(g.79752559C>G),为点突变,其父母该位点均为正常基因型,该突变可能影响mRNA的编辑加工和表达,见图1。检索人类基因突变数据库(HGMD)数据库无该位点突变的报道,根据美国医学遗传学与基因组学学会(ACMG)的《ACMG 遗传变异分类标准与指南》,该变异被解释为“可能致病性变异”[4]。患儿最终被诊断为CHUJANS。随访10个月,患儿经HCG治疗后睾丸体积较前略有增大。
讨论
CHUJANS是新近发现的一种罕见肥胖综合征,属常染色体显性遗传病,可在婴儿期发病,其临床特征是整体发育迟缓、行为异常、智力障碍、肥胖和畸形[5]。CHUJANS与PHIP基因的杂合突变相关,PHIP结构的改变是CHUJANS表型的主要驱动因素,该基因存在于6q14染色体上,最初在智力障碍患者中被发现。大多数患有CHUJANS的患者无家族史,其变异出现在生殖细胞或胎儿发育过程早期。PHIP通过可变剪接编码3种不同蛋白质,包括PHIP1、PHIP及NDRP,其中PHIP1是长约1 821氨基酸的蛋白质,又称为DDB1和CUL4相关因子14(DCAF14),是泛素连接酶复合物的多种底物受体之一,因此PHIP基因突变可通过破坏泛素连接酶途径致病,参与信号转导、蛋白质运输、染色体修饰、有丝分裂等过程[6-9]。PHIP基因还可与胰岛素受体底物1的pleckstrin同源结构域相互作用参与调节胰腺细胞的生长和胰岛素介导的信號转导,如胰岛素介导的有丝分裂、基因表达及葡萄糖的转运,尤其是可调节骨骼肌细胞中胰岛素受体刺激的葡萄糖转运蛋白4(GLUT4)易位,GLUT4易位通过自身构象改变将葡萄糖摄入细胞内,在维持血糖稳定及能量代谢中具有重要作用,因此PHIP基因或编码蛋白的功能被中断,致使患者出现肥胖[7, 10]。
目前,在ClinVar数据库中(2022年10月3日访问),关于PHIP基因有20个为移码突变,67个为错义突变,32个为无义突变,9个为剪接突变。依据ACMG的分类标准,52个为良性多态性变异,10个为疑似良性变异,58个为意义未明变异,42个为疑似致病变异,55个为已知致病突变。
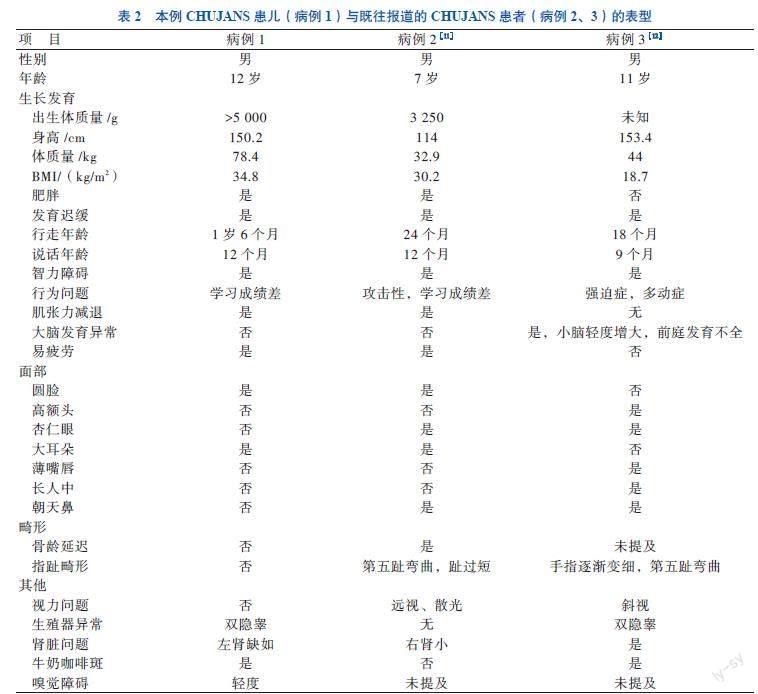
CHUJANS的临床表现多种多样,除特征性临床表现外,还可同时累及多个系统,表现出不同症状。突变体表达使基因组中复制不足的区域暴露,基因组的不稳定性增加,复制叉的稳定性与一系列临床表现相关。CHUJANS患儿神经系统表现可出现多种行为障碍,包括注意缺陷障碍、刻板行为、自闭症、智力发育落后等,颅脑MRI表现多为正常,部分可以见到脑回和脑沟略有扩大,可见增宽的透明隔腔。皮肤表现可见牛奶咖啡斑,患儿往往还存在特殊面容,包括高额头、杏仁眼、厚耳垂、长人中、宽鼻尖、上眦赘皮等。本文报道的患儿的临床表型与既往Kaur[11]、Jansen等[12]报道的PHIP基因突变个体具有相似表型,见表2。本例患儿基因突变位点位于PHIPg.79752559C>G,该基因位点突变可改变RNA前体剪接方式,该基因突变所致严重程度不一,可能与突变位点存在相关性[13]。研究显示,跨越6q14.1区域的微缺失患者也具有相似的临床表现,但存在一些额外的表现,考虑可能与6q14中相邻基因的缺失有关[14-15]。
CHUJANS的治疗大多采用对症支持治疗。对于肥胖的管理应按照标准指南进行,目的是在不影响患儿身体健康及生长发育的状态下,减少能量摄入、增加能量消耗,使体脂减少并接近于正常状态。常见的治疗方式包括:生活方式干预、心理行为干预、药物治疗及减重手术。发育迟缓者可在医师指导下给予基因重组人生长激素替代治疗,定期监测身高、体质量 、性发育状态等。对于存在智力障碍及行为问题的患儿,可于专业的儿童康复中心行系统化治疗,对于部分肌张力减退的患儿,也可在康复治疗师的帮助下,指导家长及患儿共同参与练习。
综上,Chung-Jansen综合征早期表现缺乏特异性,在目前的高生活水平状态下,儿童肥胖已成为当今社会紧迫的公共健康问题之一,需及时合理地进行诊断评估及治疗,对于存在不明原因肥胖,尤其是合并发育迟缓、智力障碍及畸形的患儿,应尽早完善基因检查,以明确诊断,早期诊断并采取综合方案治疗有助于改善患儿预后。
参 考 文 献
[1] Tirado-Class N, Hathaway C, Chung W K, et al. PHIP variants associated with Chung-Jansen syndrome disrupt replication fork stability and genome integrity. Cold Spring Harb Mol Case Stud, 2022, 8(5): a006212.
[2] de Ligt J, Willemsen M H, van Bon B W M, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med, 2012, 367(20): 1921-1929.
[3] 宋传路, 梁树夏. 全外显子组测序诊断1例罕见Chung-Jansen综合征. 中国优生与遗传杂志, 2022, 30(12): 2258-2261.
[4] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015, 17(5): 405-424.
[5] Webster E, Cho M T, Alexander N, et al. De novo PHIP-predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features. Cold Spring Harb Mol Case Stud, 2016, 2(6): a001172.
[6] Townsend A, Lora G, Engel J, et al. DCAF14 promotes stalled fork stability to maintain genome integrity. Cell Rep, 2021, 34(4): 108669.
[7] Farhang-Fallah J, Yin X, Trentin G, et al. Cloning and characterization of PHIP, a novel insulin receptor substrate-1 pleckstrin homology DomainInteracting protein. J Biol Chem, 2000, 275(51): 40492-40497.
[8] Kato H, Chen S, Kiyama H, et al. Identification of a novel WD repeat-containing gene predominantly expressed in developing and regenerating neurons. J Biochem, 2000, 128(6): 923-932.
[9] Craddock K E, Okur V, Wilson A, et al. Clinical and genetic characterization of individuals with predicted deleterious PHIP variants. Cold Spring Harb Mol Case Stud, 2019, 5(4): a004200.
[10] Marenne G, Hendricks A E, Perdikari A, et al. Exome Sequencing Identifies Genes and Gene Sets Contributing to Severe Childhood Obesity, Linking PHIP Variants to Repressed POMC Transcription. Cell Metab, 2020 , 31(6): 1107-1119.e12.
[11] Kaur H, Panigrahi I. Chung–Jansen syndrome with obesity. Obes Res Clin Pract, 2021, 15(3): 303-305.
[12] Jansen S, Hoischen A, Coe B P, et al. A genotype-first approach identifies an intellectual disability-overweight syndrome caused by PHIP haploinsufficiency. Eur J Hum Genet, 2018, 26(1):54-63.
[13] Bezrookove V, Nosrati M, Miller J R 3rd, et al. Role of elevated PHIP copy number as a prognostic and progression marker for cutaneous melanoma. Clin Cancer Res, 2018, 24(17): 4119-4125.
[14] Becker K, Di Donato N, Holder-Espinasse M, et al. De novo microdeletions of chromosome 6q14.1-q14.3 and 6q12.1-q14.1 in two patients with intellectual disability - further delineation of the 6q14 microdeletion syndrome and review of the literature. Eur J Med Genet, 2012, 55(8/9): 490-497.
[15] Wentzel C, Lynch S A, Stattin E L, et al. Interstitial deletions at 6q14.1-q15 associated with obesity, developmental delay and a distinct clinical phenotype. Mol Syndromol, 2010, 1(2): 75-81.
(收稿日期:2023-04-16)
(本文編辑:洪悦民)
猜你喜欢
少儿美术·书法版(2021年12期)2021-10-24
少儿美术·书法版(2021年9期)2021-10-20
少儿美术·书法版(2021年7期)2021-10-20
少儿美术·书法版(2021年11期)2021-10-20
少儿美术·书法版(2021年10期)2021-10-20
少儿美术·书法版(2021年8期)2021-10-20
少儿美术(2019年8期)2019-12-14
少儿美术(2019年3期)2019-12-14
杂文选刊(2016年7期)2016-08-02
小天使·一年级语数英综合(2016年6期)2016-05-14
