
钾-氯离子协同转运体2信号通路与神经病理性疼痛的关系
2023-11-24 13:51夏阳阳
赣南医学院学报 2023年9期
苏 珍,夏阳阳,黄 诚
(1. 赣南医学院康复学院;2. 赣南医学院基础医学院;3. 赣南医学院公共卫生与健康管理学院,江西 赣州 341000)
神经病理性疼痛(Neuropathic pain, NP)是由周围或中枢神经系统原发或继发性损伤引起的疼痛,包括自发性疼痛、持续性疼痛、阵发性疼痛和诱发性疼痛[1-2]。电针是一种广泛用于缓解临床疼痛的有效治疗方式,尽管在作用效果方面得到了充分证实,但其镇痛机制仍有许多未知。电针起源于我国传统医学针灸疗法,将其施加于体表特殊部位可起到通经止痛、舒畅经血,调理脏腑等作用[3]。同时,具有不良反应小、安全便捷、操作简单、容易推广等特点,广泛应用于临床疼痛的治疗,其疗效亦得到了国内外学者的认可[4],但其具体作用路径尚未阐明。研究表明,钾-氯离子协同转运体2(K+-Clcotransporter 2, KCC2)主要存在于神经系统,在致痛的产生和持续过程中表达的水平和功能不同,对于维持神经系统的稳态和疼痛调节有重要作用[5]。本文拟对KCC2 信号通路与神经病理性疼痛的关系进行综述,为进一步探讨电针是否通过KCC2 信号通路调控神经病理性疼痛提供理论依据。
1 KCC2的结构和功能
KCC2 由溶质载体家族12 成员5(SLC12A5)基因编码而成,它是受钾离子浓度梯度和Na+-K+-ATP泵的影响而驱动Cl-排出到细胞外的K-Cl共转运蛋白体[6],分为KCC2a 和KCC2b 两种亚型,它们的生理功能相似,在一定程度上可以互补[7]。如图1 所示,KCC2 是由含12 个跨膜结构域(Transmembrane domain,TMD)及胞内N 末端和C 末端的氨基酸/多胺/有机阳离子超家族盘曲折叠而成,主要结构特征表现为跨膜螺旋5(Transmembrane domain 5,TM5)和跨膜螺旋6(Transmembrane domain 6,TM6)之间的大环所形成的细胞外的结构域(Extra-cellular domain,ECD),以及跨膜螺旋12(Transmembrane domain 12,TM12)的C 末端结构域(C-terminal domain,CTD)[8-10]。CTD 可通过调节N 末端肽段的自身抑制状态维持KCC2 活性,结合在胞质腔中的N 末端肽段通过两种方式发挥这种抑制作用[11]。首先,N 末端肽段可阻断细胞内的溶剂与向内构象中的底物结合。其次,有研究表明,比较KCC1 在向内和向外构象中的结构,当封闭细胞内门时,KCC1 的TM8 螺旋、TM6b 和TM7 之间的细胞内环以及TM2 和TM3之间的短螺旋会协调一致向内运动而阻塞细胞质出口[12]。
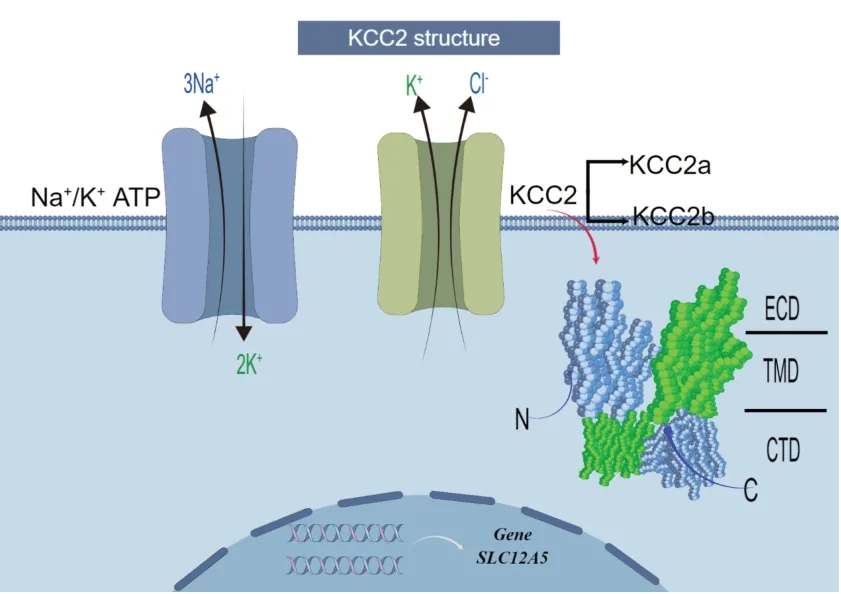
图1 KCC2分子结构
KCC2 是维持氯离子水平稳定的主要功能转运体,可驱动成熟哺乳动物神经元内Cl-浓度低于其电化学平衡电位,从而使γ-氨基丁酸A 型受体(γ-aminobutyric acid type A receptor,GABAAR)超极化并增强突触后抑制水平[13]。GABAAR 是抑制性神经递质GABA 的受体,也是中枢神经系统Cl-配体门控通道,它对细胞功能的影响作用取决于膜电位细胞内外的Cl-浓度,并尽可能维持其相对恒定。当发生神经病理性疼痛时,GABAAR 通道打开使得KCC2 功能低下而引起氯平衡失调,Cl-向细胞内流入的浓度会大于KCC2 介导的Cl-向外排出的浓度,促使细胞内高Cl-浓度,继而激活GABAAR 并导致去极化[14]。同时,有研究报道,增强GABAAR 活化可以降低KCC2 的扩散系数,从而提高膜两侧浓度的稳定性;反之,应用GABAAR 抑制剂可增加KCC2 的扩散系数,这会降低细胞膜表面的稳定性和活性[15]。此外,KCC2 和NKCC1 两种共转运蛋白参与GABA 信号传导过程是神经系统疾病重要的治疗靶点,如神经病理性疼痛、痉挛、癫痫等[7]。KCC2 在神经元树突棘形态的发生中也发挥着重要作用,并且与诸多神经系统疾病有关,深入探究其作用机制可为治疗神经系统疾病提供新策略。
2 KCC2的信号通路传递
脑源性神经营养因子(Brain derived neurotrophic factor,BDNF)是研究最多的神经营养因子之一,它是一种可以保护神经生长活性的蛋白质,原肌球蛋白相关激酶B(Tyrosine Kinase receptor B, TrkB)是BDNF 高亲和力的受体,BDNF 与TrkB 结合以及激活可调节大脑神经功能。嘌呤受体P2X 配体门控离子通道4(Recombinant purinergic receptor P2X ligand gated ion channel 4, P2X4)是一种ATP门控阳离子通道受体,其表达变化与神经疼痛信号的传递有关,ATP激活小胶质细胞上的P2X4受体后促进BDNF 释放,而BDNF 可通过激活神经元上的TrkB 受体,引起KCC2下调,进而改变神经元的兴奋性。BDNF-TrkB-KCC2 是参与神经病理性疼痛发生和调控的主要信号传导通路,且一些研究认为P2X4-BDNF-KCC2轴适用于特定神经病理性疼痛机制的调节,KCC2 作用于目标区域的状态又可通过反馈给GABA 能神经元和5-HT 能神经元改变下行兴奋或抑制。此外,PLCγ1 和RAS/MAPK 途径可改变KCC2 mRNA 水平。除了一些信号途径,还有支架蛋白、磷酸化状态、蛋白激酶、转录因子、促炎因子和离子浓度等也可改变KCC2 水平,最终影响痛觉信号的传递。
KCC2 的调控依赖于BDNF 对TrkB 的激活,BDNF 的过表达可显著提高体内KCC2 mRNA 表达水平[16],而TrkB 受体的基因敲除会显著抑制KCC2 mRNA 表达[17],这说明激活BDNF-TrKB 可能是通过下调KCC2 引起痛觉过敏。对于P2X4-BDNF-KCC2轴,激活P2X4 依赖性的小胶质细胞可促进脊髓背角BDNF的释放,进而下调神经元的KCC2转运蛋白体,促进神经病理性疼痛的发生发展[18]。此外,KCC2 富集在GABA 能神经元和谷氨酸能神经元的突触附近,其作用机制可通过反馈途径调节神经元对下行疼痛信号传递的抑制作用。进一步研究表明神经损伤可降低脊髓背角KCC2 的功能,并将中缝大核5-HT的下行抑制转变为下行兴奋,进而阻断5-HT 作用于局部脊髓GABA 能和甘氨酸能神经元的下行抑制和镇痛作用[19]。如图2 所示,GABA 能神经元的突触传递还可以通过阻断TNFR1-S1PR2-CCL2-CCR2-BDNF-TrkB 通路的任一节点使其达到正常化,进而改善由神经炎症引发的GABA 能神经元传递变化相关的疾病[20]。
有研究[21-22]发现BDNF 与TrkB 的结合可导致TrkB 受体内酪氨酸残基的自磷酸化,并激活磷脂酶Cγ1-钙调蛋白依赖性蛋白激酶途径、磷酸肌醇激酶3 途径和RAS/MAPK 途径,而磷脂酶Cγ1 途径可抑制KCC2 mRNA 表达,RAS/MAPK 途径则促进KCC2 mRNA 转录(图2)。亦有研究发现,抑制性突触主要支架蛋白表达量对成熟海马神经元KCC2 介导的氯离子调节有着重要作用[23],这提示支架蛋白可能是KCC2 的新型关键调节因子。谷氨酸能神经元[24]和GABA能神经元[15]的突触传递可以通过侧向聚集或扩散方式对KCC2功能施以调节,使得KCC2介导的氯化物排出减少或增加,这一级联反应涉及KCC2 的C 末端结构域中特定丝氨酸和苏氨酸残基的磷酸化和去磷酸化[15,25]。KCC2 的扩散还可通过其表面活性的翻译后修饰进行调控,这涉及不含赖氨酸(K)(WNK)及其效应物——丝氨酸/苏氨酸激酶39(SPAK)(STE20/SPS1 相关,以及富含脯氨酸-丙氨酸的激酶)/OSR1[26],这一途径是依据氯离子浓度敏感性来调节它的活动,是KCC2 的重要调节器。N-甲基-D-天冬氨酸是一种离子型谷氨酸受体,其活性受KCC2 的磷酸化状态调节[27],在没有信号级联反应的情况下,KCC2 的功能也可通过直接磷酸化进行信号调节,如KCC2的C 端残基第940丝氨酸可受N-甲基-D-天冬氨酸磷酸化的影响,从而改变神经细胞对其表达量的稳定调控。KCC2 功能的发挥取决于神经细胞内外钾离子浓度差,而星形胶质细胞对维持细胞外低钾离子浓度非常重要,这表明星形胶质细胞参与了KCC2 的功能调节以及神经病理性疼痛的发展。通过刺激蛋白激酶或G蛋白偶联受体也可调节KCC2 活性,如星形胶质细胞分泌的IL-6 通过活化蛋白激酶A 来促进BDNF 的表达,以及激活TrkB 受体下调KCC2 的表达[28-29]。组蛋白去乙酰化酶(Histone deacetylase 2,HDAC2)抑制剂对周围神经损伤引起的机械痛和热痛敏具有显著的抑制作用,通过上调KCC2 途径以减轻神经病理性疼痛[30]。与此同时,在坐骨神经慢性压迫模型(Chronic constriction injury model,CCI)诱导的神经病理性疼痛中可以观察到MAPK磷酸化和NF-κB核易位上调,它们位于促炎因子合成和分泌的上游,继而影响KCC2 的功能[31]。此外,星形胶质细胞中锌离子浓度升高也同样可下调KCC2 表达[32]。以上证据表明KCC2 信号通路在神经病理性疼痛中发挥重要作用。
综上所述,在神经病理性疼痛中主要通过一些信号级联反应来调节KCC2 的功能,这可反馈给抑制性中间神经元对其下行疼痛的兴奋或抑制作用;另一些途径可调控KCC2 翻译表达水平以保持痛觉系统的平衡。此外,KCC2 本身的磷酸化状态、神经细胞表达的蛋白、释放的促炎因子、蛋白激酶的激活以及细胞内外的离子浓度差等各种反应均可调节KCC2 表达,并在神经病理性疼痛的信号传递中发挥重要作用。
3 中枢神经系统KCC2与神经病理性疼痛
脑干中缝大核、中脑导水管周围灰质、延髓头端腹内侧核和脊髓背角是伤害性信息传递过程中下行调控转换的枢纽,参与调控神经病理性疼痛。这些传导连接来自更高脑部的综合输入形成BDNF/ TrkB-KCC2 信号通路,并直接投射到脊髓背角相应痛觉传递神经元[33]。脊髓背角神经元内的氯化物水平对于神经病理性疼痛的信息传递至关重要,因为神经元里氯化物浓度决定了GABA 能神经元和甘氨酸能神经元对神经信息传递是否具有抑制作用[34]。氯化物失衡是神经元突触抑制受到损害的重要机制[35],当神经受到损伤时,神经元内的氯化物浓度会升高,引发氯平衡的失调(如KCC2功能低下)进而诱导神经病理性疼痛,若恢复氯离子平衡调节可逆转由SNI诱导的神经病理性疼痛[36]。大量证据表明,对KCC2 表达量的调控在神经炎症和神经病理性疼痛中起着重要作用[37]。神经元的KCC2 是维持氯化物浓度稳定的重要共转运蛋白体。有研究报道,周围神经损伤后促使脊髓背角浅层神经元的KCC2 表达量减少,从而影响疼痛感知,即对外周刺激表现为超敏反应以及不再对脊髓突触的阴离子电流产生抑制反应[5]。另有研究表明,卵巢切除术可以通过改变脊髓背角KCC2 蛋白表达水平来调节神经病理性疼痛对镇痛剂的敏感性[38]。我们已知α7 烟碱型乙酰胆碱受体(α7 nicotinic acethlcholine receptor,α7nAChR)激活后可通过抑制小胶质细胞的活化以及BDNF/TrkB/KCC2 信号通路调节神经病理性疼痛[39]。脊髓的小胶质细胞可分为M1 促炎型和M2 抗炎型,两种极化状态共同维持神经系统的稳态。M1 型小胶质细胞可负性调控并上调促炎因子的分泌表达量,如IL-6、iNOS、CD16/32、TNF-α、IL-1β 和CD68 等,这些物质可进一步加重神经系统损伤[40-41]。而M2 型小胶质细胞可上调抗炎因子的分泌量,如IL-10、Arg-1、CD206 以及转化生长因子等,这些物质对神经元起到消炎镇痛的保护作用[40-41]。最近研究也证实,尽管神经免疫信号传导在性别当中存在少许差别,但氯化物平衡调节在神经病理性疼痛中同样适用于雄性和雌性啮齿动物[42]。其他研究也揭示了脊髓背角细胞多样性以及KCC2 是如何在疼痛中参与信号通路的形成[43],但目前仍有许多概念未能研究清楚,比如,当不清楚什么样的脊髓细胞处于氯离子失调状态,就不能确定这些细胞的峰值是如何受到影响的,也就无法判断氯离子失调是如何影响信号传导的,以及这些细胞的通路功能改变是如何参与神经病理性疼痛的形成[44]。
脊髓背角的KCC2 在不同的神经病理性疼痛模型的突触传递模式也各不相同,这种疼痛机制是由脊髓背角的氯化物平衡失调所致[45]。这说明通过改变KCC2 的功能可能有利于缓解周围神经损伤引发的神经病理性疼痛。有研究显示,亚甲基芳香化合物CLP257 作为KCC2 激活剂虽然不能改变它的蛋白活性,但是可以增强GABAAR 的活性[46],从而产生一定的镇痛效果。KCC2 表达降低可导致共转运蛋白介导的氯离子外流,进而不能维持细胞内低氯化物浓度[19,42],因此,氯化物失衡可能是SNI 诱导神经病理性疼痛的关键机制。总之,脊髓背角的KCC2 下调是神经细胞氯化物失调的重要原因,我们应当聚焦KCC2 功能的调控,这有助于将神经病理性疼痛问题具体化,而KCC2 可能是治疗慢性疼痛的绝佳药物靶点。
4 外周神经系统KCC2与神经病理性疼痛
在伤害性痛觉信息传递过程中,须通过外周神经系统参与初级感觉神经元的传导和调控,与更高位的神经元形成突触连接。由于实验条件的限制,大多数以DRG 感觉神经元作为外周神经系统疼痛的研究对象。DRG 感觉神经元可表达大量高活性的KCC1和KCC3,但KCC2的表达少且活性低,甚至难以检测[47]。因此,目前对DRG 神经元KCC2 的表达研究存在一定的不确定性[48]。尽管如此,也有学者通过PCR 和免疫荧光技术检测到DRG 中KCC2 mRNA 和蛋白质弱表达[49]。DRG 神经元的氯离子浓度主要由细胞膜上的转运蛋白NKCC1、KCC2 以及ATP 酶共同维持,KCC2 将细胞内的氯离子泵至细胞外,导致胞内低氯离子浓度和负平衡电位,而NKCC1 将氯离子从外部转运到细胞质,从而使氯离子电位更加去极化[50]。此外,炎症介质会降低DRG中KCC2 的表达,与NKCC1 共同作用导致细胞内氯离子水平升高和痛觉神经元兴奋性增强[51]。因此,致炎因子可以通过影响DRG 神经元的氯离子浓度增加GABA 去极化,而NKCC1 可缓解神经病理性疼痛的热痛和机械痛反应[52]。这些都提示了DRG 中KCC2 维持氯离子浓度稳定和电导对于神经病理性疼痛的重要性。有研究[50]表明,疼痛会改变初级传入感受器的氯离子通道、转运蛋白和稳态,反之,氯离子通道以及转运蛋白的活性改变则会影响疼痛感知。通过将携带KCC2 的病毒注射到DRG 神经元,发现可以逆转GABA介导的电位去极化,并减轻神经病理性疼痛的痛超敏反应[53]。但也要考虑到啮齿动物中氯离子介导的痛觉稳态与人类的稳态机制并不完全相同[54]。近年来,在中枢敏化过程中,发现脊髓背角痛觉传入神经纤维的兴奋性增加,会在低阈值刺激下激活DRG[22]。尽管KCC2 调控神经病理性疼痛的作用机制主要发生在脊髓背角,但DRG 神经元也参与了伤害性刺激的信号传递,仍然是不可忽视的重要环节,需要进一步加以研究。
5 小结与展望
由于神经病理性疼痛的机制复杂,且目前没有针对病因的特效药,临床应用的镇痛药仅能暂时起到一定的缓解作用,而频繁使用不仅易出现耐受且不良反应较多。电针(Electroacupuncture, EA)由中国传统医学针灸发展而来,它能够促进大鼠正中神经的再生和神经干细胞的生长,还可通过刺激中枢和外周神经的活性因子来减轻神经病理性疼痛,加之不良反应少,因此被广泛用于治疗慢性疼痛。许多患者服用一定剂量的镇痛药物而达不到缓解疼痛的预期效果,而电针恰好属于安全实惠且耐受良好的保守治疗方式。然而,电针对神经病理性疼痛患者的治疗效果主要是根据其临床表现,较少涉及内在机制的评估和分析,为此,探究电针缓解神经病理性疼痛的作用机理,可为临床治疗慢性疼痛寻找新的理论依据和有价值的治疗靶点。研究表明电针镇痛机制与KCC2 功能的增强有关。因此,立足于研究增强KCC2 的表达途径,可能有助于揭开电针镇痛机制的神秘面纱,为广大疼痛患者带来福音。
猜你喜欢
遵义医科大学学报(2023年6期)2023-06-30
基层中医药(2018年11期)2019-01-31
中成药(2018年12期)2018-12-29
科普童话·神秘大侦探(2017年4期)2017-04-06
厦门理工学院学报(2016年1期)2016-12-01
浙江大学学报(工学版)(2016年2期)2016-06-05
中国病理生理杂志(2015年8期)2015-12-21
汽车生活(2015年6期)2015-05-30
建筑材料学报(2015年3期)2015-02-28
中国应用生理学杂志(2014年3期)2014-01-22
