
基于ISSR标记的多花黑麦草附球菌遗传多样性分析
2023-11-22 04:21:02徐志婷许玲玲薛龙海
西南农业学报 2023年9期
徐志婷,许玲玲, 2,薛龙海
(1. 兰州大学草地微生物研究中心,兰州 730000;2. 中国林业科学研究院,北京 100080)
【研究意义】多花黑麦草(Loliummultiflorum)是禾本科黑麦草属的一种优良牧草,我国主要种植在长江流域以南的地区。其产量高、适口性好、生长速度快,因此作为南方畜牧业首选的牧草之一[1]。除此之外,多花黑麦草也可进行青贮,弥补我国南方冬春季青饲草的不足[2]。附球菌属(Epicoccum)是重要的植物病原真菌,其归属于子囊菌门(Ascomycota)、亚隔孢壳科(Didymellaceae),可侵染农作物、林木、经济作物等,造成植物叶斑、幼苗和果实腐烂[3-8],发病严重时,可造成作物产量下降20%~35%[3]。【前人研究进展】Epicoccum的寄主范围很广泛,可引发多种植物病症。其属内的黑附球菌(E.nigrum) 对小麦(Triticumaestivum)[9]、甜瓜(Cucumismelo)[10-11]和玉米(Zeamays)[12]等植物都表现出一定的致病力。 据报道,黑附球菌在津巴布韦引起棉花(Gossypiumhirsutum)[13]病害,在日本造成水稻颖壳上的病斑[14];冬青被黑附球菌侵染后,会造成叶片脱落、果实腐烂和变小,最终坏死。Gupta等[15]报道了一种附球菌,引起了重要的豆科栽培牧草埃及车轴草(Trifoliumalexandrinum)的叶斑病。在我国,也从茶树(Camelliasinensis)上分离得到了3种不同的附球菌(E.camelliae、E.layuense和E.sorghinum),叶部病害的发生严重影响了茶叶的产量和品质[4,16]。附球菌在多花黑麦草上普遍发生,但目前国内外对多花黑麦草上的附球菌属病原真菌研究极少,仅在部分禾草上有相关研究。Wiewióra等[17]曾从黑麦草种子中分离到黑附球菌E.nigrum,在巴西曾报道过E.sorghinum引起雀稗属(Paspalumguenoarum)植物叶斑病,导致50%叶面积被病斑覆盖[18];在美国,从鸭茅(Dactylisglomerata)种子中分离到E.nigrum[19];E.plurivorum自新西兰狗尾草(Setariasp.)分离得到[20];在澳大利亚发现E.viticis会对澳大利亚的非洲须芒草(Andropogongayanus)造成一定程度的危害[4]。在我国,由E.sorghinum引起的马唐(Digitariasanguinalis)褐斑病的田间叶片发病率高达70%以上[21];燕麦(Avenasativa)作为饲用作物在我国北方地区和西北地区大面积种植,由E.layuense引发褐斑病的发生极大地影响了其产量和品质[8]。分子标记技术根据DNA序列差异直观地反映遗传多样性,包括以Southern杂交技术为核心的分子标记技术、限制性片段长度多态性(RFLP)、以PCR技术为核心的标记技术,如随机扩增多态性(RAPD)、扩增片段长度多态性(AFLP)、简单重复序列区间(ISSR)、简单重复序列(SSR)、序列相关扩增多态性(SRAP)以及DNA芯片为核心的三代分子标记技术[22]。其中,ISSR标记操作简单、稳定性好、多态性强,因此在植物病原菌的分类、亲缘关系和遗传多样性等领域被广泛应用。比如烟草镰刀菌根腐病菌[23]、苹果腐烂病[24]、烟草赤星病[25]、桑褐斑壳丰孢菌[26]和桑毛色二孢根腐病[27]。【本研究切入点】很多研究已表明附球菌可以导致多种作物病害,但国内外对附球菌遗传多样性的相关研究相对匮乏,尤其是多花黑麦草上的病原附球菌。本研究基于ISSR标记研究多花黑麦草附球菌遗传多样性,为多花黑麦草附球菌病害的病原多样性研究提供理论意义。【拟解决的关键问题】以贵州、云南、重庆和四川等地的黑麦草叶片上分离的37株附球菌为研究对象,通过采用ISSR分子标记分析5个不同地理来源Epicoccum菌株的遗传多样性,构建系统发育聚类图并进行主成分分析,探究病原菌菌株的遗传分化规律是否与菌株的地理来源有关,以期从分子水平解析附球菌属真菌的种内是否存在与地理相关的遗传分化。
1 材料与方法
1.1 试验材料
1.1.1 供试菌株 2021—2022年从贵州花溪、云南曲靖、四川崇州和重庆南川、合川5个地区采集典型多花黑麦草叶斑病病样,经分离和纯化后,进行形态学观察和DNA测序,通过引物ITS、TUB2、RPB2和LSU,筛选了93株附球菌菌株,从中选出37株代表性菌株,菌株的详细信息见表1。
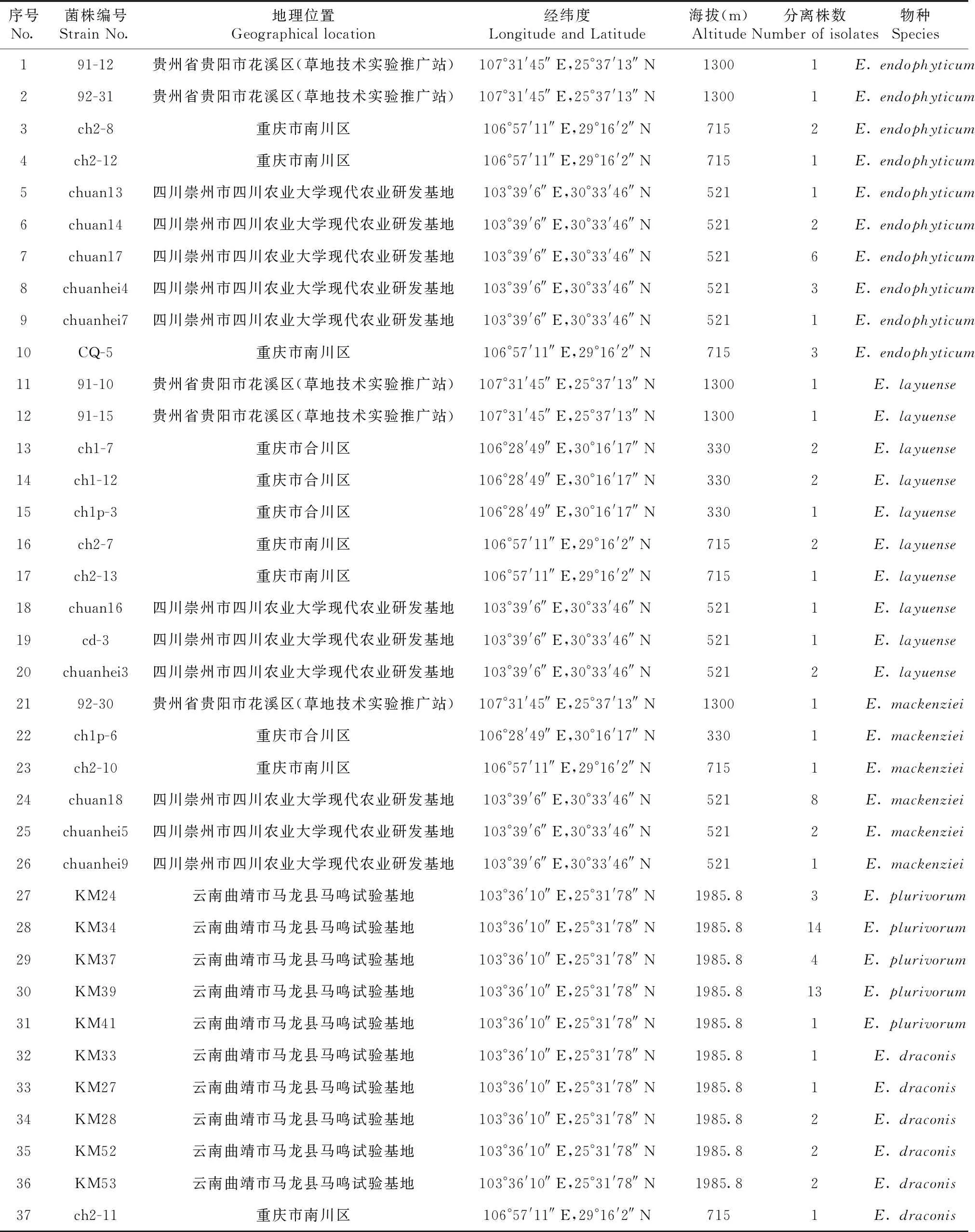
表1 供试菌株信息
1.2 方法
1.2.1 附球菌的分离 采用组织分离法对病原菌进行分离。采集发病的多花黑麦草叶片,在病健交界处用无菌剪刀剪取5 mm×5 mm的小块,先在75%酒精中消毒30 s,再用无菌水冲洗3次,置于灭菌滤纸上风干后,平铺至PDA培养基,置于25 ℃培养箱中黑暗培养。待叶片组织长出少量菌丝时,挑取菌落边缘的菌丝至PDA培养基上进行多次纯化。
1.2.2 DNA提取 采用Omega真菌 DNA 提取试剂盒提取病原真菌DNA,提取具体方法参照试剂盒说明书进行,DNA浓度的检测通过Promega-NanoDrop超微量分光光度计(Thermo公司,美国)进行,使用1.0%琼脂糖凝胶电泳检查质量,合格的DNA(>30 ηg/μL)进行稀释后置于-20 ℃条件下备用。
1.2.3 ISSR-PCR反应 35条引物选自加拿大哥伦比亚大学公布的100条ISSR引物序列,合成由生工生物工程(上海)股份有限公司完成。初步的筛选主要是使用35条引物对不同地区的8株菌株DNA模板进行扩增,以筛选出条带多。多态性及重复性好的引物,再用得到的引物对37株附球菌菌株进行ISSR-PCR扩增分析。PCR扩增反应体系包括:12.5 μL 2×SanTaqPCR Mix, 9.5 μL ddH2O,上下引物各1 μL (10 μmol/L), DNA模板1~2 μL。PCR反应程序:95 ℃预变性3 min;95 ℃变性30 s,退火温度50~60 ℃(退火温度根据引物设定),退火时间30 s,72 ℃延伸50 s,35个循环;72 ℃延伸10 min。PCR扩增产物通过1.5%琼脂糖凝胶电泳检测之后,利用凝胶成像系统拍照。
1.2.4 ISSR数据分析 从加拿大哥伦比亚大学公布的ISSR引物序列中随机选择35条引物先对8株菌株的DNA进行扩增,从中选择扩增条带数量多、条带清晰的引物,之后对所有供试菌株的DNA进行扩增,对特定大小的扩增片段,根据条带的有无分别记为1和0,对所得二进制矩阵进行统计分析。采用NTSYSpc(Version 2.10e)对不同菌株之间遗传相似系数进行计算,通过构建系统发育聚类图进行聚类相关分析;利用PopGene(Version 1.32)对不同地理来源菌株的遗传分化水平进行分析[25];将二进制矩阵整理成GenAIEx6.51软件可识别数据格式,计算遗传距离,基于遗传距离进行主成分分析(PCoA)。
2 结果与分析
2.1 引物筛选和ISSR扩增
通过使用35条ISSR引物对8个DNA模板进行扩增,筛选出12条引物,这些引物具有较好的多态性。进一步筛选后,得到6条重复性较好。多态性条带较多的引物(表2)。筛选出的6条引物共扩增出62条谱带,各引物的扩增条带数为8~15,扩增片段大小为 250~1000 bp,所有引物均表现出较高的扩增多态性,引物ISSR4扩增的条带数最多(图1)。

M:DNA标记;1:92-31;2:chuan18;3:chuanhei4;4:chuanhei7;5:ch1-12;6:chuanhei3;7:chuan17;8:CQ-5;9:cd-3;10:ch2-13;11:KM34;12:ch2-8;13:chuan16;14:92-30;15:91-15;16:ch1p-3;17:KM37;18:KM24;19:KM41;20:KM39;21:KM27;22:KM28;23:ch2-7;24:ch1-7;25:chuanhei5;26:91-10;27:KM53;28:ch2-12;29:KM52;30:91-12;31:KM33;32:ch2-11;33:ch2-10;34:chuan14;35:chuanhei9;36:chuan13;37:ch1p-6。图1 引物ISSR4的扩增结果Fig.1 Amplifying result of the primer ISSR4

表2 6条ISSR引物扩增的条带数
2.2 ISSR标记的菌株聚类分析
由图2可知,不同地理来源的附球菌菌株遗传相似系数在0.3100~0.9300。当遗传相似系数为 0.4180时,37株附球菌被划分为5个类群,这5个类群分别代表不同的附球菌菌种,说明ISSR类群划分与菌种分类之间存在一定的相关性。其次除E.plurivorum种群之外,E.layuense、E.mackenziei、E.draconis和E.endophyticum种群包含来自不同地理来源的菌株。E.layuense菌株在重庆、贵州、云南和成都4个地区都有分布,E.plurivorum菌株主要来自云南地区。聚类分析结果显示,E.endophyticum菌群和E.mackenziei菌群亲缘关系较近,E.layuense菌群、E.draconis菌群和E.plurivorum菌群亲缘关系较近。E.layuense种群内的遗传分化与地理来源未发现显著相关性。

图2 37株菌株的UPGMA聚类分析Fig.2 UPGMA cluster analysis of 37 Epicoccum isolates
2.3 ISSR标记的菌株居群遗传分化
利用PopGene进行计算,得出各居群的遗传距离(D)和Neis遗传一致度(IN)。研究结果(表3)表明,5个居群的遗传一致度介于0.8863~0.9478,平均值为0.9168;遗传距离在0. 0536~0.1207,平均0.0871。5个居群中,曲靖居群(KM-)的Epicoccum菌株群体和合川居群(ch1-)的Epicoccum菌株群体的遗传距离最长(D为0.1207),遗传一致度最低(IN为0.8863),表示这2个居群间的遗传分化程度最高,亲缘关系较远;而花溪(91-;92-)居群的Epicoccum菌株群体和崇州居群(川-;川黑-)的Epicoccum菌株群体遗传一致度最高(IN为0.9478),遗传距离最短(D为 0.0536),表示这2个居群间的遗传分化程度最低,亲缘关系较近。
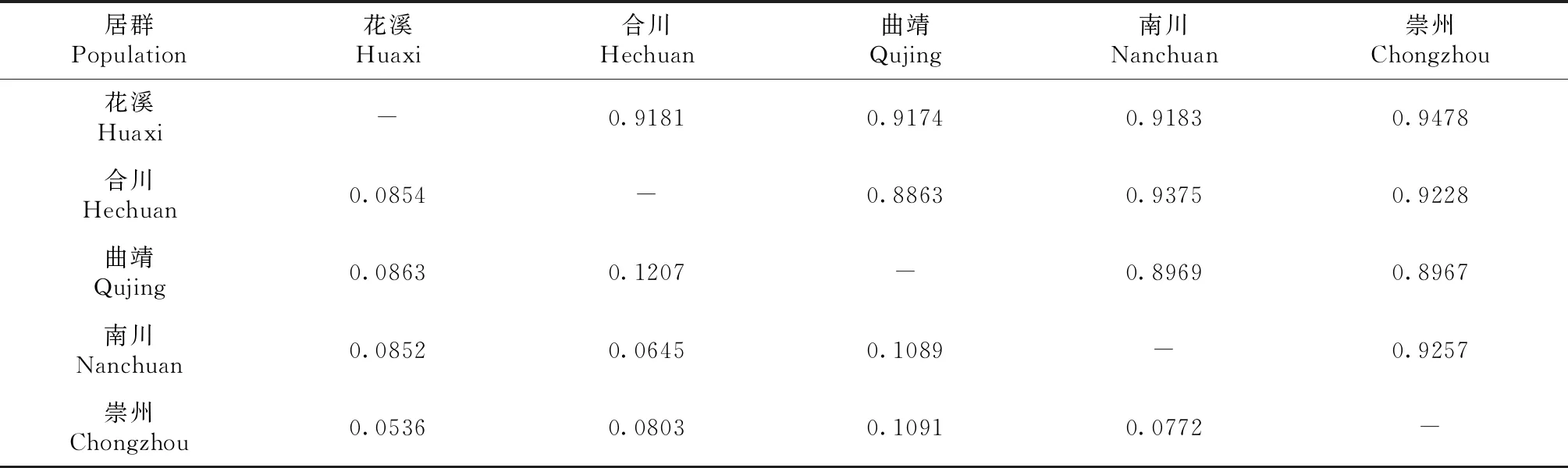
表3 Epicoccum菌株5个居群的Neis遗传一致度和遗传距离
2.4 ISSR标记的菌株群体多样性
由表4可知,通过PopGene计算5个群体的遗传多样性参数,得出5个居群的平均多态位点数(Pl)为51.00个,平均多态位点百分率(Ppl)为80.95%,平均观察等位基因数(Na)为1.8095,平均有效等位基因数(Ne)为1.5407,平均基因多样性指数(H)为0.3121,平均Shannon’s 信息指数(I)为0.4606。具体而言,崇州居群的Pl最多,至58.00个,Ppl为92.06%;合川居群和曲靖居群的Pl和Ppl最低。居群内的遗传多样性以崇州居群的菌株最高,其I为0.5306,曲靖居群的I最低,为0.4293。
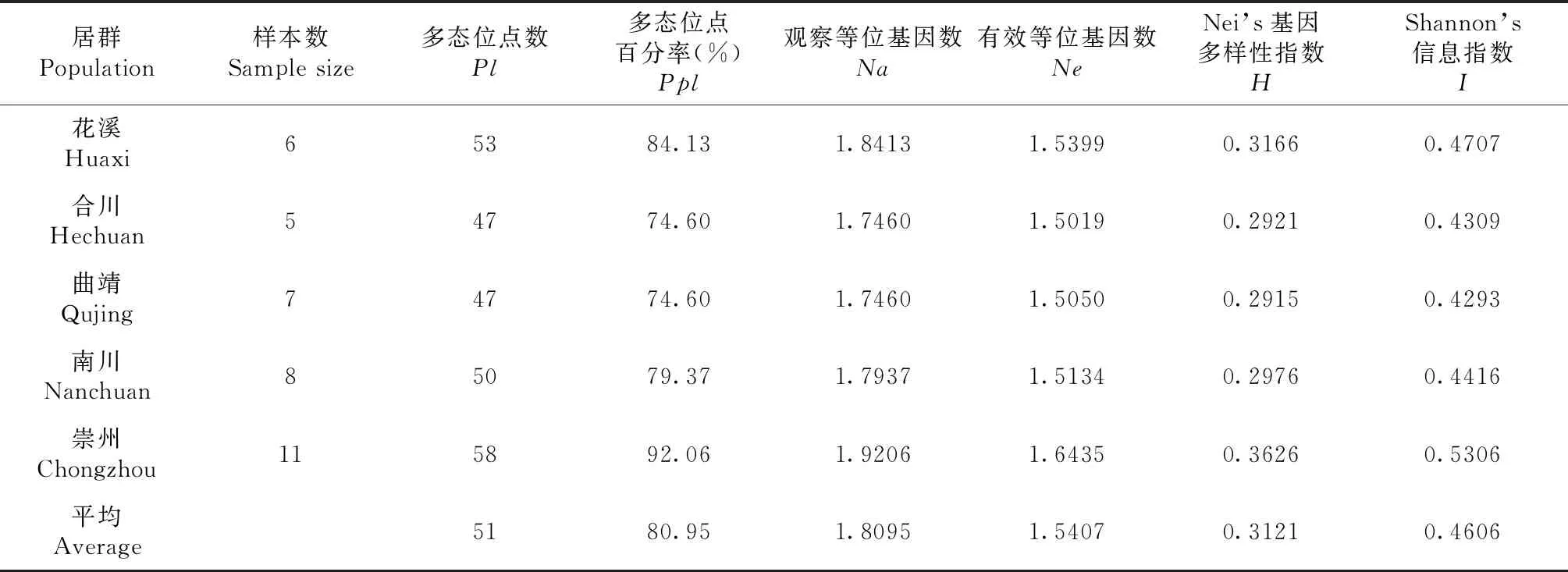
表4 Epicoccum菌株5个居群的遗传多样性水平
由表5可知,37株供试菌株在种水平上的Na为2.0000,Ne为1.6305,H为0.3628,I为0.5388,菌株的总杂合度Ht平均为0.3583,种水平遗传杂合度Hs为0.3121,遗传分化系数Gst为0.1289,基因流Nm为3.3794。通过分析表4~5的结果可知,居群间的H和I低于居群内的H和I,表明居群内的遗传分化程度相较于居群间较高。

表5 37株Epicoccum菌株的遗传多样性水平
2.5 PCoA分析
PCoA分析是基于ISSR数据获得的遗传距离得出。在PCoA分析中,不同地理来源的菌株随机分布(图3),未观察到个体的分组,居群(pop1和pop4)相交但又有差异,在遗传多样性和菌株的地理起源之间没有发现相关性。

pop1:贵州花溪;pop2:重庆合川;pop3:云南曲靖;pop4:重庆南川;pop5:四川崇州。pop1:Huaxi;pop2:Hechuan;pop3:Qujing;pop4:Nanchuan;pop5:Chongzhou.图3 基于ISSR显示Epicoccum分布的主成分分析(PCoA)Fig.3 Distribution of Epicoccum isolates revealed by principal coordinates analysis (PCoA) based on ISSR
3 讨 论
DNA分析技术在真菌的遗传多样性方面被广泛应用,其中ISSR分子标记技术稳定性高、多态性丰富,在Fusarium、Valsa、Alternata、Phloeospora和Lasiodiplodia等病原菌的遗传多样性研究中具有显著优势。本文基于ISSR标记方法分析5个不同地理来源的群体遗传多样性,扩增得到多态性条带63条,多态率达100%。
研究发现,部分真菌菌株的遗传多样性与菌株的地理来源有一定联系,但大部分真菌的遗传多样性与菌株的地理来源间不具相关性。例如,龙珑[28]以叶点霉属和茎点霉属30个分离菌株为供试材料,应用ISSR分子标记手段对两属真菌进行系统分类学研究,结果发现叶点霉属的分类与地理来源之间未表现出相关性;王兴红[29]利用ISSR分子标记技术分析来自国内柑橘主产区和国外收集的135株叶点霉属(Phyllostictaspp.)菌株的遗传多样性,未发现叶点霉的遗传变异与地理分布明显相关。在本研究中,曲靖类群和合川类群的地理距离最远,遗传一致度最低,E.plurivorum类群的地理来源相同,聚类分析中菌株聚在同一分支,但不同地理来源的E.layuense菌株聚在一支,即同一地区的菌株可聚到不同的分支。来源于西南4省的37株附球菌菌株被划分为5个居群,PCoA结果显示,菌株的划分与地理来源无关,不同地理来源的菌株没有分组,结果表明菌株的遗传分化水平与地理来源没有显著相关性,这与上述研究结果相似。
本研究ISSR分子标记数据显示菌株间的遗传分化水平显著低于居群间的遗传分化水平,这可能与不同地区的地理、生态和气候环境有关,崇州居群的遗传多样性最高,可能与该地种植品种较多有关,主要有川农1号、安第斯、杰威、长江2号等。当遗传变异数(Gst)值低于0.15时,遗传分化主要体现在群体间[20],基因交流值(Nm)为3.3794,进一步说明群体间的遗传变异程度相较于群体内高。虽然PopGene结果分析发现不同居群间遗传变异程度较高,但总体而言,群体间的遗传相似度较高,这可能是由于5个地区种植的多花黑麦草品种较为相似。由于本研究中分离到的菌株数量有限,可能无法全面反映多花黑麦草上附球菌种群内的遗传变异,因此需进一步收集大量菌株,以研究其种群内的遗传变异情况,为多花黑麦草叶斑病的防控提供理论依据。
4 结 论
多花黑麦草上分离到的附球菌菌株多态性高,ISSR类群划分与菌种分类结果一致,居群间的遗传多样性高于菌株间的遗传多样性,菌株的地理来源与其遗传多样性之间不存在相关性。
猜你喜欢
奥秘(创新大赛)(2022年8期)2022-09-14 05:44:42
河北科技师范学院学报(2022年2期)2022-08-26 08:55:24
中国果业信息(2021年10期)2021-12-07 04:06:06
浙江中医药大学学报(2021年6期)2021-07-12 03:05:14
养殖与饲料(2020年6期)2020-02-18 16:01:26
中国种业(2019年8期)2019-08-26 08:41:18
草地学报(2018年5期)2018-11-07 02:25:00
系统工程与电子技术(2016年2期)2016-04-16 05:16:53
中国光学(2015年1期)2015-06-06 18:30:20
海岸工程(2014年4期)2014-02-27 12:51:28
