
染色体核型分析深度学习方法综述
2023-11-22 01:18罗纯龙赵屹
中国图象图形学报 2023年11期
罗纯龙,赵屹
1.中国科学院计算技术研究所泛在计算系统研究中心,北京 100080;2.中国科学院大学,北京 100049
0 引言
染色体是遗传物质的载体,是细胞核中最重要的组成部分。因此,研究染色体成为科研人员在细胞水平上认识遗传规律的桥梁。染色体核型分析技术恰好是一种基于视觉特征的研究染色体的细胞遗传学分析技术,并广泛应用于各个领域。林娟等人(2020)研究发现染色体核型分析有益于为慢性粒细胞白血病(chronic myelogenous leukemia,CML)的诊断、病情发展和预后提供重要信息。郑国兵等人(2020)通过分析180 例产妇产前诊断情况,认为染色体核型分析是一种有效的产前诊断方法,能够有效降低新生儿出生缺陷发生风险。此外,窦笑菊(2012)展望了染色体核型分析对植物学研究的推动和促进。
健康人群分裂中期细胞中一般包含22 对常染色体和1 对性染色体,共计46 条染色体。核型分析技术将采集的细胞经培养、制片和染色后,通过高倍光学显微镜获取细胞分裂中期图像,如图1(a)所示。然后,各种基于数字图像处理的计算机辅助分析系统(如CytoVision、Ikaros 和ASI HiBand)(Xiao等,2020)辅助细胞遗传学家人工完成染色体计数、分离粘连和重叠的染色体等。最终根据染色体形态结构特征将所有染色体按照人类细胞遗传学国际命名体制(international system for human cytogenetic nomenclature,ISCN)进行排列,形成核型图像,如图1(b)所示。
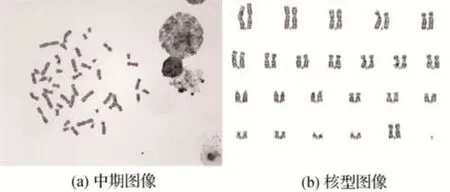
图1 中期图像以及核型图像Fig.1 Metaphase image and karyotype image((a)metaphase image;(b)karyotype image)
随着人们不断拓展染色体核型分析应用领域,特别是在产前诊断中的广泛应用,核型分析需求日益上涨。但是,传统染色体核型分析过程冗长乏味且耗时。以染色体计数任务为例,一位经验丰富的细胞遗传学家按规范计数一位病人的所有染色体约需15 min(Xiao 等,2020)。这给高质量、高效率的染色体核型分析带来挑战。一种可行的解决方案是采用染色体核型自动化分析,在关键且耗时的分析步骤中以自动化方法代替人工分析,细胞遗传学家仅需校验自动化分析结果,出具诊断报告。染色体核型自动化分析不仅可以极大地提高核型分析效率,减轻人员负担,同时还能保持核型分析结果的一致性,避免人为因素导致的性能波动,便于核型分析技术进一步推广。
染色体核型自动化分析方法早期主要利用传统数字图像处理方法和机器学习方法(Abid 和Hamami,2018;Lin 等,2020b),例如基于数字图像处理的染色体计数方法、基于染色体中轴特征的多层感知机分类模型以及一些基于阈值和几何特征的染色体分割方法等。但是,一方面染色体非刚性结构特点导致其极易弯曲变形和粘连重叠;另一方面实验环境操作手法等不可控因素导致染色体成像质量、形态结构和条带水平等方面差异较大。这使得人工提取的形态特征和基于阈值和规则的方法泛化能力较弱,性能较差,难以应对复杂多变的临床场景,辅助作用有限。此外,其他任务如异常生成等问题传统方法难以解决。深度学习技术凭借其数据驱动特点和强大的学习能力在计算机视觉特别是医学影像处理领域得到快速发展。由于该技术可以从大规模形态结构各异的染色体数据中自动学习与任务相关的特征而无需人工干预,基于深度学习的染色体核型自动分析方法表现出明显优于传统方法的性能和泛化能力,已成为研究热点。但是,尽管基于深度学习的染色体核型自动化分析方法研究已经取得了一定成果,相关综述性工作依然存在不足:
1)国内一些与染色体核型分析相关的综述性工作存在缺陷。陈少洁等人(2021)仅讨论了基于深度学习的染色体分割研究工作。邱俊玮和孙频捷(2021)则仅选择了少数热点方向的工作进行论述,缺乏对基于深度学习的染色体核型自动分析工作的全面讨论。
2)染色体核型分析任务与临床紧密结合,因此方法会受到任务目标、数据量和数据标注等多方因素限制,复杂多变类型众多,需要梳理研究方法类型,理清研究脉络。
3)深度学习模型在自然图像领域的飞速发展导致当前研究工作多集中于染色体分割和分类任务,其余分析任务尚未引起广泛关注,需要以综述的形式详细介绍其他染色体核型分析任务和可行的解决方案。
本文系统性地梳理了近10 年基于深度学习的染色体核型自动化分析关键任务问题范式、算法研究现状、尚需解决的难点挑战,最终提出了后续发展方向。主要包括:1)阐述了经典目标检测方法及提出的基于框检测的染色体计数方法;2)从语义分割范式和实例分割范式两个方面出发描述了染色体分割方法;3)强调了染色体簇分类的必要性,综述了基于卷积神经网络(convolutional neural network,CNN)的染色体簇分类方法并强调了统一簇类别标准的重要性;4)从染色体预处理任务出发,分别归纳了细胞分裂中期图像降噪和染色体矫直两种预处理任务的可行解决方案;5)归纳整理了基于简单 CNN 结构、经典 CNN 结构、特征对比、图像预处理、全局和局部特征融合和复杂策略的 6 种染色体分类方法;6)针对染色体异常梳理了与异常发现和异常生成任务相关的深度学习方法;7)收集整理了公开可获取的染色体核型分析相关数据集;8)简单总结了6 种染色体核型分析任务的算法研究成果,还讨论了当前的研究热点以及亟待解决的问题,提出了染色体核型自动化分析领域未来可能的发展方向,为读者提供参考和帮助。
1 染色体计数
1.1 问题归纳
染色体异常中有一类是染色体数目异常。如李静等人(2020)所报告的21-三体综合征就是一种因存在3条21号染色体而造成生长发育迟缓的染色体数目异常。所以,染色体计数往往是人染色体核型分析的第一步。尽管可以通过完整的核型分析达到染色体计数目的,但为了避免误差累积和过度消耗计算资源,仍然需要独立的自动化染色体计数方法。已有的基于深度学习的染色体计数方法主要将其转化为目标检测任务,需要从细胞分裂中期图像中预测全部染色体,如图2所示。

图2 基于框检测的染色体计数方法预测结果Fig.2 Prediction results of chromosome counting algorithm based on bounding boxes detection
1.2 基于框检测的染色体计数方法
Xiao等人(2019)首先发现了染色体计数任务存在的两个难点:1)自相似性问题(self-similarity problem),即染色体的一部分或头尾相接的两条染色体被识别为一条染色体;2)严重重叠的染色体簇会对基于交并比(intersection over union,IoU)度量的后处理目标检测模型产生干扰。
为了解决以上两个问题,基于Faster R-CNN(faster region-based convolutional network)(Ren 等,2017)框架(如图3 所示),Xiao 等人(2019)提出了DeepACE 模型。该模型首先优化了第1 阶段正负锚框(anchor)采样方法。根据存在自相似性问题的假阳性目标对应的锚框分布规律将负例锚框(negative anchors)细分为难负例锚框(hard negative anchors)和简单负例锚框(easy negative anchors)。随后通过分层采样维持样本平衡,即难负例锚框采样(hard negative anchors sampling,HNAS)方法。对于严重重叠导致的问题,DeepACE 模型总结染色体几何分布规律设计了5种模板掩码(template mask),并结合弱监督模板模块(weakly-supervised template module)和基于距离的损失函数为每个候选提议(candidate proposal)提取了模板特征(template feature)。这些模板特征将与交并比度量一起在新的特征引导的非极大值抑制(feature-guided non-maximum suppression,Feature-guided NMS)后处理方法中发挥作用,尽可能保留不同模板特征代表的染色体实例,避免假阴性结果。

图3 Faster R-CNN模型结构(Ren等,2017)Fig.3 The architecture of Faster R-CNN(Ren et al.,2017)
但是,DeepACE 方法并没有考虑到严重遮挡和重叠对染色体定位造成的影响,因此提出了升级版DeepACEv2(Xiao 等,2020)。除优化特征提取网络为ResNet(residual network)(He 等,2016;Lin 等,2017),DeepACEv2 还简化了模板模块(template module),并设计了嵌入引导的非极大值抑制(embedding-guided NMS)方法替代原有后处理过程。最后,相比Wang等人(2018)提出的排斥损失(repulsion loss),DeepACEv2 为了避免惩罚重叠染色体的固有区域,提出了对不精确定位更加敏感的截断归一化排斥损失函数(truncated normalized repulsion loss,TNRL)来缓解严重的类内重叠导致的定位精度下降问题。为了进一步提高模型的推理速度,Kang等人(2022)将轻量级网络MobileNetv3(Howard 等,2019)作为Faster R-CNN 框架的特征提取网络,以实现推理加速。
1.3 总结
综上所述,现有方法均将染色体计数转化为目标检测任务。计数任务往往缺乏染色体类别信息,导致模型对染色体长度和条带模式不敏感,容易造成自相似性问题;另一方面,重叠的染色体对预测轴对称边界框造成干扰。此外,两阶段目标检测模型的推理加速也是研究人员重点关注的方向。现有方法一方面从检测网络出发,在锚框采样方法、检测头结构、后处理过程和边界框回归损失函数等方面针对染色体形态结构特征进行优化,取得了较好的效果;另一方面则从特征提取网络出发,通过替换轻量级骨干网络实现模型推理加速。但是,非刚性的染色体导致人工设计的模板尚不能完全包含所有可能的染色体形态。严重重叠染色体之间的干扰即使通过唯一标识的特征予以缓解,但不可避免的低检测分数仍无法满足临床高置信度检测要求。下一阶段,研究人员一方面可以尝试引入旋转矩形框标注,通过预测旋转边界框,规避轴对称矩形框中存在的干扰问题,但需要重点关注染色体多向性导致的角度预测挑战;另一方面,基于关键点的染色体计数方法也有较大的潜力。通过预测具有重要语义信息的关键点如着丝粒和两侧端点等,也可以实现染色体计数目的。染色体计数方法的简要总结如表1所示。

表1 染色体计数方法简要总结Tabel 1 A brief summary of the chromosome counting methods
2 染色体分割
2.1 问题归纳
染色体异常中另一类是染色体结构异常,从细胞分裂中期图像中分割出完整独立的染色体实例是核型分析中重要的一步。但是,因为非刚性染色体容易弯曲变形,细胞分裂中期图像中经常出现染色体粘连或重叠的现象,这成为染色体分割任务面临的重大挑战。为了完成染色体分割任务,已有的基于深度学习的方法根据可以简单划分为基于实例分割和基于语义分割的染色体分割方法,如图4 所示。前者需要精确识别每条染色体实例,所以基于Mask R-CNN(He 等,2017)的模型被该类方法广泛采用。后者仅需识别同类像素,所以研究人员提出了许多基于U-Net(Ronneberger 等,2015)及其变种的染色体分割方法。
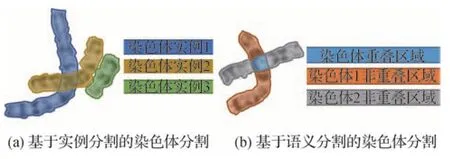
图4 基于实例分割和语义分割的染色体分割Fig.4 Illustration of chromosome segmentation based on instance segmentation and semantic segmentation((a)chromosome segmentaiton based on instance segmentation;(b)chromosome segmentation based on semantic segmentation)
2.2 基于语义分割网络的染色体分割
Hu 等人(2017)提出了基于U-Net(Ronneberger等,2015)的染色体分割模型。该方法将染色体重叠区域单独作为一个类别,形成了背景、染色体1 非重叠区域、染色体2非重叠区域和染色体重叠区域4类分割任务。该方法考虑到染色体图像大小和染色体数目与形态都较为单一,因此减少了模型深度,避免网络过拟合,提高染色体分割性能。最后将模型预测的非重叠区域和重叠区域组合形成完整染色体。该方法首次尝试引入深度学习解决染色体分割问题,相对非深度学习方法适用范围更广。Saleh 等人(2019)同样提出了基于U-Net 的染色体分割模型解决两两重叠染色体的分割问题。受到训练数据规模限制,模型改进了U-Net 架构,相比原始架构减少了层数和通道数以避免模型过拟合。此外,还引入测试时增强策略(test time augmentation,TTA)(Wang等,2019)来对重叠染色体图像进行数据增强,扩大训练数据多样性,进一步缓解过拟合问题,提高模型性能。但是,不同类别的染色体大小迥异,导致固定的感受野无法适配染色体大小,而直接裁剪或缩放存在信息丢失的风险。所以,Wang 等人(2021b)提出了一种基于U-Net 架构的自适应感受野多尺度网络(adaptive receptive field multi-scale network,ARMS Net)。ARMS Net 应用残差路径(residual path)模块取代了原始U-Net 框架中的跳跃连接,充分利用编码器模块中丢失的空间信息来补偿编码器和解码器之间的语义差距。ARMS Net 还提出了自适应多空洞卷积(adaptive multi atrous convolution,AMAC)和自适应相同步长池化(adaptive same stride pooling,ASSP)组成自适应多尺度特征提取器,实现自适应提取融合多尺度特征。对于像素级分类任务中存在的不平衡问题,ARMS Net对正负样本的倍数进行平滑处理当作损失权重。Mei 等人(2022)则从另一角度思考,提出了对抗性多尺度特征学习(adversarial multiscale feature learning,AMFL)框架来提高重叠染色体分割的准确性和适应性。AMFL 将重叠染色体图像分割转化为图像到图像的翻译(image-to-image translation)任务,通过条件生成对抗网络(conditional generative adversarial network,cGAN)(Isola 等,2017)来惩罚生成的类别决策图和源图像之间的差异,推动生成器为分割任务生成更高置信度的决策结果。AMFL 选择UNet++(Zhou 等,2020)网络作为cGAN 的生成器,通过结构中不同深度的密集跳跃连接完成多尺度特征融合,满足不同尺度目标分割需要。最后,AMFL 将Lovasz-softmax(Berman 等,2018)作为分割损失实现更高分割性能。Song 等人(2022a)则提出了一种称为Compact Seg-UNet 的新型卷积神经网络。模型以SegNet(Badrinarayanan等,2017)为主体框架,不仅移除数个卷积层以减少过拟合风险,还融入了U-Net 的跳跃连接以期弥补下采样丢失的信息。编码器中的最大池化层会保留池化索引(pooling indices),帮助解码器中的上采样算子更好地恢复特征图尺寸。
但是,以上方法仅能解决两条染色体重叠的问题,当面对3 条或更多染色体重叠时需要新的方法。面对这一挑战,Cao 等人(2020)提出了两阶段ChromSeg(chromosome segmentation)模型。模型的第1 阶段将UNet++框架扩展为双分支输出框架,利用浅层和相邻的空间特征将背景与染色体实例分离开来,同时聚合多层次语义特征来预测交叉重叠区域。在两个分支中,ChromSeg 模型应用混合权重焦点损失(mixed-weight focal loss,MWF)来平衡正负样本训练。模型第2 阶段应用交叉分区算法(crossingpartition algorithm)从候选交叉重叠区域中提取染色体实例。该算法以重叠区域为中心,通过延长到非重叠区域中心的射线寻找到对应的另一块非重叠区域,得到完整的染色体实例。但是,ChromSeg 方法除了交叉重叠区域预测错误和非重叠区域配对错误外,最大的不足是仅能解决有限场景的区域匹配,无法解决多个重叠区域紧密聚集导致的匹配错误。为了弥补这一缺陷,Liu 等人(2022b)尝试应用启发式算法从数学和几何的角度来改进匹配过程。该研究提出了两点假设,一是重叠区域对应的非重叠区域数量最多为4 个;二是正确匹配的非重叠区域的中心坐标连线的交点与重叠区域的中心坐标的欧氏距离更近。基于以上两点假设,在UNet++分割出重叠区域与非重叠区域后,该研究使用Removeredundant 算法通过比较欧氏距离过滤掉不属于当前重叠区域的非重叠区域,最终得到匹配正确的染色体实例。
遗憾的是,以上方法均需要将完整的染色体分区域预测,致使拼接时可能因为分割精度的问题出现空洞等瑕疵。Bai 等人(2020)希望过滤掉细胞核等杂质后,专注于预测完整的染色体。所以,该研究提出了一种基于YOLOv3(you only look once v3)(Redmon 和Farhadi,2018)和U-Net 优化组合的染色体分割方法。首先利用U-Net 去除细胞分裂中期图像中的细胞核等杂质,然后应用YOLOv3 模型完成染色体检测并沿边界框裁剪得到染色体切片。由于切片中可能存在重叠或粘连的染色体片段,所以再次应用U-Net 模型分割切片,得到无需拼接的染色体实例。
2.3 基于实例分割网络的染色体分割
相较于基于语义分割的染色体分割方法,基于实例分割的染色体分割方法对于输入数据的限制更小。林成创等人(2020)认为临床任务中遇到的复杂染色体簇存在多条染色体同时粘连和重叠的现象,因此基于路径增强网络(path aggregation network,PANet)(Liu 等,2018)模型,提出了数量统计分割路径增强网络(amount segmentation PANet,AS-PANet)模型完成重叠染色体簇实例分割。AS-PANet 除继承原有预测分支外,还接入了染色体簇实例数目预测分支,将其作为额外的监督信号提高分割性能。但是,由于训练数据量较少,分割性能仍有提升空间。Huang 等人(2022)针对这一问题,设计了一个染色体实例标注数据集增强算法(chromosome instance labeled dataset augmentation,CILA)来提升模型的泛化性能。因为染色体是非刚性目标,染色体簇的形态和方向具有多样性,所以CILA 通过随机翻转和随机平移旋转扩增成多个角度的样本供Mask R-CNN(He 等,2017)模型训练,缓解小数据集中方向偏好造成的泛化误差。
但是,为了尽可能实现染色体自动化分割,需要设计将细胞分裂中期图像作为原始输入的方法。Huang 等人(2021)认为尽管染色体丰富的形变使得基于几何的方法难以大规模应用,但深度学习所需要的大规模高质量数据集也会提高成本,因此提出了一种新的结合几何特征和Mask R-CNN 的染色体分割方法。该方法首先应用基于几何连通性的等值轮廓查找算法(iso-valued contour finding algorithm)将细胞分裂中期图像划分为仅包含染色体实例和染色体簇的切片(splices),然后提取染色体切片的11个几何特征,利用逻辑回归算法找到所有的染色体簇切片,最后利用Mask R-CNN模型分割染色体簇得到染色体实例。所以,这一方法仅需标注染色体簇,大幅降低数据标注要求和成本。Pijackova等人(2022)选择应用U-Net 模型去除细胞分裂中期图像中的细胞核和杂质,完成图像降噪。然后应用Otsu 阈值算法和骨架方法提取候选目标的中轴之后,即可通过计算端点数目确定候选目标是染色体实例还是染色体簇。对于染色体簇,Pijackova 等人(2022)进一步利用Mask R-CNN 完成染色体簇分割,并借助TTA技术减少模型预测的假阴性和假阳性结果。然而,同样是几何特征结合Mask R-CNN 的染色体分割方法,Chang 等人(2021)则先行通过Mask R-CNN 模型分割出染色体实例和染色体簇,对于后者,再通过凸包法和最小包围矩形(minimum bounding rectangle,MBR)来判断真假染色体簇,最后通过交叉点完成染色体簇分割。但是实验结果发现,首尾相连的染色体簇仍会对这一方法造成挑战。
以上方法结合几何特征和深度学习,虽然实现了从细胞分裂中期图像分割染色体实例的目的,但是多阶段非端到端的方式流程复杂,容易积累误差。因此,冯涛等人(2020)尝试将染色体方向信息融入模型中,设计了基于Mask R-CNN 的端到端染色体实例分割方法Mask Oriented R-CNN。方法在原有的3 个分支的基础上新增了预测有向边界框(oriented bounding box,OBB)的分支,以更紧密的边界框完成染色体精确定位。针对有向边界框,提出了新的角度加权交并比(angle-weighted intersectionover-union,AwIoU)度量来代替IoU 度量。在掩码分支中,还实现了有向卷积通路(oriented convolutional path,OCP)结构,使得不同朝向的染色体实例可以在不同的卷积路径上进行训练,减少了染色体之间粘连重叠对掩码预测的干扰。除了方向信息,Wang等人(2021c)还尝试加入染色体端点信息作为监督信号,提出了增强旋转Mask R-CNN(enhanced rotated Mask R-CNN)模型,利用多种监督信息来提高模型性能。该论文认为,狭长的染色体可以通过两侧端点完成大致定位,因此该模型在引入旋转边界框检测分支的基础上,还引入了一个端点检测分支,通过端点检测任务为染色体定位提供更丰富的信息。Liu等人(2022a)观察到实例分割模型一般分类置信度较高但分割性能较差,提出了基于回归修正(regression correction)的染色体实例分割网络。该方法在不额外增加分支的情况下,利用边界框回归损失得到与定位精度强相关的回归置信度,同时还预测与分割精度相关的分割掩码交并比,两者进一步修正分类置信度。其次,该研究还提出了更符合染色体空间分布规律的基于实例掩码交并比的NMS 算法代替基于边界框交并比的NMS 算法。最后,基于回归修正的染色体实例分割网络还设计了K-交并比损失(K-IoU loss)函数,通过分割面积赋予不同权重,提高模型对错误分割的敏感性。
2.4 总结
综上所述,粘连重叠的染色体始终是染色体分割任务首先需要解决的难题。与自然图像分割任务不同的是,染色体分割任务需要将重叠区域同时分配给所有关联染色体。所以,现有染色体分割工作均重点关注重叠区域或降低染色体重叠对分割模型性能的影响。已有工作可以简单分为基于语义分割的染色体分割模型和基于实例分割的染色体分割模型。
基于语义分割的方法一般需要先将图像像素分为背景、染色体重叠区域和染色体非重叠区域,然后将不同区域组装成完整染色体。但是这类方法面临如下主要难点:1)需要精确区分染色体重叠区域和非重叠区域;2)需要解决不同尺度染色体对语义分割模型的影响;3)需要正确配对染色体重叠区域和非重叠区域,重点是多条染色体重叠情况。针对这些难点问题,研究人员优化了U-Net 模型结构、提出了多尺度特征融合框架和模块、设计并优化了基于几何的匹配算法。但是,因为数据标注和算法特点,需要将完整染色体分区域预测拼接,容易造成空洞等瑕疵。
基于实例分割的方法会直接识别每条染色体所在区域并完成像素分类,但是这类方法面临的主要问题是染色体多边形标注数据不足。相当一部分研究工作将目标直接指向重叠染色体分割或提前通过几何方法从中期图像中分离出重叠染色体减少数据标注需求,再通过设计新的数据扩增方法、增加额外监督信号等方法优化基于实例分割的方法。但这种多阶段方法容易累积误差,所以另一部分研究人员直接在细胞分裂中期图像中完成端到端的染色体分割,并对经典的Mask R-CNN模型结构做出了大量优化,设计了基于方向信息的方法、端点信息监督的方法和回归修正方法等,提出了一批新的预测分支、交并比度量方法和损失函数。但是,缺乏类型丰富的高质量分割标注数据仍是制约性能进一步提升的重要因素。
接下来,为分割模型替换性能更加强劲的骨干网络、基于领域知识引入更多辅助监督信号、通过传统几何方法降低问题求解难度,甚至利用自监督学习范式降低对于高质量分割标注数据的需求都是染色体分割领域可以进一步深入的研究方向。染色体分割方法的简要总结如表2所示。

表2 染色体分割方法简要总结Tabel 2 A brief summary of the chromosome segmentation methods
3 染色体簇分类
3.1 问题归纳
正如第2 节所讨论的,相当一部分染色体分割算法应用范围有限,仅能完成特定类型的染色体簇分割任务。所以,判断染色体簇类型并选择合适的分割算法是自动化染色体核型分析重要环节。当前,针对染色体簇分类的研究工作较少,对于簇的类别定义也存在差异。
3.2 基于卷积神经网络的染色体簇分类
一种染色体簇标注规则是依据染色体簇中重叠的染色体数目赋予不同标签。例如Somasundaram(2019)将染色体簇类别划分为touching、one overlapping、two overlapping 和multiple overlapping 共4 种类别,然后提出了一个基于卷积神经网络的两阶段分类方法。第1 阶段由新设计的卷积神经网络完成染色体簇分类;第2 阶段更进一步通过形态学操作识别切割线来得到染色体实例,并训练了结构与第1阶段相同的分类网络完成正常—异常染色体分类任务。
但是,这一染色体簇标记规则忽略了复杂染色体簇中可能混合了粘连染色体和重叠染色体。因此,Lin 等人(2021)将染色体簇类别划分为instance、touching、overlapping 和touching-overlapping 共4 种类别。模型在ResNeXt 模型(Xie 等,2017)基础上设计了9 层的神经网络代替原有的输出层。新的输出层在接收到骨干网络输出的特征图后,首先由平均池化层和最大池化层组成的混合池化层提取全局特征和局部特征。这些特征经过平滑层(flatten layer)变形后输入到由若干线性层(linear layer)、修正线性单元(rectified linear unit,ReLU)、批归一化(batch normalization,BN)层和dropout 层组成的神经网络得到预测结果,以此来提高模型泛化能力。此外,该研究不仅将弱监督学习(weakly-supervised learning,WSL)预训练权重(Mahajan等,2018)作为初始权重,还通过整合One cycle leanring(Smith,2018)和Discriminative learning rate(You 等,2017)两种训练策略实现模型参数微调,在较短的时间内使模型收敛至较高水平。
3.3 总结
综上所述,染色体簇分类是完成自动化核型分析的关键步骤,但染色体簇分类任务还处于早期阶段,尚未引起大规模关注。当前染色体簇分类任务面临的主要问题是没有统一的染色体簇类别定义和大规模高质量的数据集,这些因素制约了染色体簇分类任务的高速发展。现有方法主要从分类模型结构出发,无论是设计轻量级专有模型还是引入超大规模自然图像数据集预训练权重都是为了在小规模染色体簇数据集上尽可能提高模型性能。但是,轻量级模型可能无法应对临床中复杂染色体簇样本,而自然图像与染色体图像之间也存在领域差异。所以,接下来可以从数据出发,除了大量收集并标注高质量染色体簇数据,还可以设计新的图像合成方法,包括图像线性插值法、基于生成式对抗网络方法和基于扩散模型方法等。在模型结构优化方面,由于辨别染色体簇类别的关键是粘连和重叠区域,所以模型显式提取关键区域特征进行细粒度分类或基于注意力机制自适应提取类别关键特征都是可以进一步研究的方案。染色体簇分类方法的简要总结如表3所示。

表3 染色体簇分类方法简要总结Tabel 3 A brief summary of the chromosome cluster classification methods
4 染色体预处理
4.1 问题归纳
染色体核型分析图像中不可避免地存在诸如细胞组织和污染杂质等噪声,这些噪声可能对小尺寸染色体产生干扰,因此针对细胞分裂中期图像的基于语义分割的降噪方法进入研究人员的视野。其次,染色体由于其非刚性特点,容易弯曲变形,对核型分析和后续疾病诊断造成障碍,所以一部分研究人员也设计了基于生成式对抗网络和基于运动补偿方法的染色体矫直方法。
4.2 细胞分裂中期图像降噪
Altinsoy 等人(2019)计划将所有G 显带染色体所在区域作为同一类别区域分割来实现细胞分裂中期图像降噪,提出了一种基于U-Net 的语义分割网络。但是受限于数据规模,该方法不仅减少了通道数量,还添加dropout 层进一步加强模型泛化性能。然而,这种端到端深度学习模型是单通道输出,对于预测结果中错误保留的非染色体区域无法进行修正。所以,Altinsoy 等人(2022)提出了一个由分割网络和分类网络组成的级联神经网络架构来完成降噪任务。该方法第1 步提出了新的分割网络,它融合了U-Net 模型“编码器—解码器”对称架构、加法前ReLU(ReLU before addition)残差单元和预激活(preactivation)残差单元,提高了模型正则化能力且更易于训练优化。新的分割网络细分为3 种语义类别输出,分别是背景预测掩码、染色体区域预测掩码和非染色体区域预测掩码。在第2 步中,该方法会对染色体和非染色体预测图进行二值化和形态学开运算,得到染色体或非染色体目标。这些目标的对象面积(object area)、凸面积(convex area)、染色体预测图上的平均像素值(average pixel value on chromosome prediciton map)和非染色体预测图上的平均像素值(average pixel value on non-chromosome prediction map)4 种特征将被输入到由全连接层组成的分类网络中,去除非染色体目标。
4.3 染色体矫直
使用几何方法矫直染色体时会存在输出边缘参差不齐或条带不连续的情况,这不利于后续分析。所以,Song 等人(2021)尝试应用图像—图像翻译的新框架,输入染色体的拉直骨架来生成具有无间断条带和更多细节的矫直染色体,并利用学习感知图像块相似度(learned perceptual image patch similarity,LPIPS)来度量差异。具体来说,该研究基于pix2pix 模型(Isola 等,2017)为每个弯曲的染色体都训练了一个单独的“图像—图像”翻译模型。该方法首先为每条染色体提取棍状骨架(stick backbone),并对每条染色体和其棍状骨架进行数据扩增形成一个数据集。模型将U-Net模型作为pix2pix 框架的生成器,预测的染色体将与骨架配对作为“假”样本,而真实染色体与骨架配对则作为“真”样本。对抗训练将使得生成器U-Net 网络获得棍状骨架与染色体实例之间的映射关系,预测得到更真实的染色体。但是这一方法需要为每个染色体实例训练一个图像翻译模型,计算量大且所需时间较长。Song 等人(2022b)提出了ViT-Patch GAN(vision transformerbased patch GAN)方法,一种基于cGAN(Mirza 和Osindero,2014)的框架将染色体矫直任务转化为运动变换任务(motion transformation task)。该方法将基于主成分分析的运动估计模型(pca-based motion estimation model,PMEM)(Siarohin 等,2021)作 为cGAN 的生成器。但是受限于矫直染色体数据集规模较小,PMEM 模型会生成不准确的矫直结果。与此同时,源图像和生成图像存在较大差异,传统判别器所用的卷积算子的远距离建模能力较弱,因此提出了ViT-Patch(Dosovitskiy 等,2021)判别器,通过对抗训练,输出的特征既包含染色体局部语义内容也含有整个染色体的之间的联系。最后,为了防止测试阶段的结果失真,ViT-Patch GAN 应用了SLmatching(size learned perceptual image patch similarity matching)(Zhang 等,2018a)方案来从数据集中选择相似大小和形状的图像。
4.4 总结
综上所述,染色体预处理方面主要由细胞分裂中期图像降噪和染色体矫直两个任务组成。对于降噪任务,面临的主要难点是如何正确识别细胞组织和杂质。已有的方法会将染色体区域作为整体,通过语义分割模型进行分离,或更进一步通过额外的分类模型去除错误的非染色体区域,在降低像素损失的同时减少噪声。但是标注数据集规模小,复杂多变的细胞组织和杂质及其与染色体的相似性仍是后续染色体降噪方法需要重点关注的问题。大量来自临床分析中间结果的人工降噪图像是天然的标签。一方面可以借助生成式对抗模型,将语义分割模型作为生成器,通过判别器识别分割降噪图像和人工降噪图像;另一方面可以将细胞组织和杂质作为特殊的数据变换,利用自监督学习范式使得语义分割模型的编码器部分对此类数据变换脱敏,从而达到降噪效果。
染色体矫直任务所面临的挑战在于如何形成边缘平滑、条带连续的拉直的染色体。已有的方法突破了常规的通过弯曲点剪切拼接的方法,提出了基于生成式对抗网络和基于运动补偿的染色体矫直方法,对于后者还引入了ViT模型增强远距离建模能力。但是这些方法在面对临床应用时,还无法证明合成染色体是否满足临床需求。所以未来需要同细胞遗传学家合作,通过人工分析诊断原始染色体和矫直染色体来验证合成矫直方法是否会对疾病诊断产生干扰。染色体预处理方法的简要总结如表4所示。

表4 染色体预处理方法简要总结Tabel 4 A brief summary of the chromosome preprocessing methods
5 染色体分类
5.1 问题归纳
为了出具临床报告,细胞遗传学家需要参照ISCN 识别每条染色体并根据模板出具核型图和诊断结果,所以确定染色体类别是核型分析过程中极为重要的一环。然而,Lejeune 等人(1960)研究发现某些染色体之间视觉特征更相似,不同类染色体差异较小;另一方面,由于染色制片时的环境制剂等不同,条带水平(分辨率)也不同,同类染色体差异较大。这些问题对染色体分类任务造成挑战。
5.2 基于简单CNN结构的分类方法
由于染色体分类数据集一般较小,一些研究工作尝试简化染色体分类网络结构。Sharma 等人(2017)首次提出了基于深度学习的染色体实例分类方法,尝试以众包(crowdsourcing)的形式从细胞分裂中期图像中分割并矫直染色体实例。如图5(a)所示,这些经过预处理的图像将由新设计的简单卷积神经网络分类。该网络由4 个包含卷积核、ReLU、dropout层和最大池化层的卷积模块以及两个全连接层和一个24 维的归一化指数函数(softmax)层组成。Zhang 等人(2018b)将卷积模块数目降低至2 个但增加了卷积层通道数来扩充模型的表达能力。如图5(b)所示,模型将第1 个卷积核大小设置为5 × 5,用来快速降低特征图分辨率,帮助减少参数并加速收敛。Hu 等人(2019)提出了一个由3 个卷积模块组成的CNN 网络,不同之处在于每个模块中的第2 个卷积核的大小均为5 × 5。如图5(c)所示,该方法同样在实验中发现Y 染色体数据不平衡导致的性能下降问题。Menaka 和Vaidyanathan(2022)提出了Chromenet 来完成染色体分类任务。如图5(d)所示,该网络统一应用3 × 3 卷积核,并在每个卷积核后首次引入批次归一化层。此外,Chromenet 模型仅将dropout 层插入最后的全连接层中,缓解密集连接导致的过拟合问题。Chromenet结构简单参数量少,但难以进一步提高染色体分类模型性能和泛化能力。
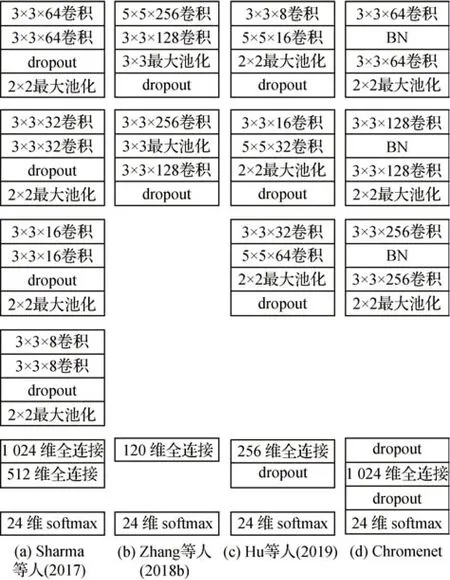
图5 基于简单CNN结构的分类网络架构Fig.5 Structure of classification network based on simple CNN((a)Sharma et al.(2017);(b)Zhang et al.(2018);(c)Hu et al.(2019);(d)Chromenet)
5.3 基于经典CNN结构的分类方法
Lin 等人(2020a)提出基于聚合残差架构(aggregated residual architecture)的染色体分类网络MixNet。该模型选择将ResNeXt 作为骨干网络并将自适应网络头(adaptive network header)作为分类器。为了加速收敛,MixNet 选择先训练自适应网络头,再对模型整体微调。Wang 等人(2021a)认为如果染色体数据来源单一将会弱化模型泛化性能,所以提出了LseNet 网络评估模型在混合数据集上性能。LseNet 网络集ResNet(He 等,2016)和SENet(squeeze-and-excitation networks)(Hu 等,2018)于一身,在ResNet50 网络中间数层中加入了Lse 模块(leaky squeeze-and-excitation block)。而为了进一步增强模型判别能力,除交叉熵损失外,模型还应用了中心损失(center loss)来聚拢类内特征。Sharma 等人(2018)则提出了残差卷积循环注意力神经网络(residual convolutional recurrent attention neural network,Res-CRANN)来完成染色体分类任务。模型首先利用ResNet50 提取染色体实例特征,随后将高为G、宽为H、通道数为K的特征图沿着通道维度连接形成数量为G、特征维度为H×K的特征序列。然后,Res-CRANN 利用长短期记忆网络(long-short term memory network,LSTM)建模染色体条带之间的长距离依赖关系。但是,由于序列长度较长,Res-CRANN 还采用了注意力机制使得分类模型更关注特征序列中与类别相关的感兴趣区域,预测结果也更加准确。
5.4 基于特征对比的分类方法
Swati 等人(2017)选择首先矫直弯曲的染色体,但不同的是该研究采用了基于中轴提取及众包的矫直(straightening via medial axis extraction and crowdsourcing,SMAC)和基于投影向量的矫直(straightening via projection vectors,SPV)两种方法。接着,设计了孪生网络(siamese network)架构,通过能量函数度量两个分支输入的低维特征之间的欧氏距离,使得同类样本在特征空间中的低维映射足够接近,不同类别样本距离更远。最后,这些样本的低维映射向量将由一个两层前馈神经网络确定具体类别。Wang 等人(2020)受到基于孪生网络的染色体分类方法的启发,提出了基于特征距离度量的染色体分类模型。该研究首先在ResNet50 的骨干网络后扩展了一个由全连接层、ReLU 和dropout 层组成的扩展块(extened block),并在训练中采用交叉熵损失函数和中心损失函数,增强模型判别能力。其次,该方法还将验证集中数据特征汇总形成24 类对应的标签特征向量(label feature vector,LFV),然后应用基于豪斯多夫距离的标签再分配策略(label redistribution strategy,LRd),通过两轮计算缩小标签空间,预测最终结果。但是该方法中LFV 来自验证集,性能受制于验证集质量。除了孪生网络的方式,Gajjar等人(2022)还尝试通过三元组损失(triplet loss),利用较少的数据训练相似性模型。该研究提出了两种方案,分别是离线三元组损失(offline triplet loss)和在线三元组损失(online triplet loss)。基于欧氏距离的三元组损失将约束网络使得锚与正例尽量靠近而远离负例,而训练得到的高维嵌入将通过多层感知机或K最近邻分类算法完成染色体分类。
5.5 基于图像预处理的分类方法
Swati 等人(2018)认为缺乏高分辨率染色体实例图像是制约染色体分类性能的关键问题,因此提出了Super-Xception 模型,通过卷积超分辨率层将低分辨率图像转换为高分辨率图像,然后由Xception(Chollet,2017)分类网络判断染色体类别。其中,卷积超分辨率层由3 个卷积—修正线性单元层组成,该模块将双三次插值到一定大小的低分辨率图像作为输入,然后学习低分辨率图像到高分辨率与低分辨率图像差值的映射。卷积超分辨率层的输出加上插值后的低分辨率图像形成了超分辨率图像,最后由Xception 分类网络判断类别。Ding 等人(2019)通过阈值处理和形态学操作的方法得到间距分散的染色体,在识别染色体轮廓后完成染色体分割。但是考虑到成像质量不稳定,该研究还加入了基于染色体核型图像频率特征的对比度增强方法,通过维纳滤波器(wiener filter)和低通滤波器(low pass filter)等方法放大高频分量,增强染色体条带特征并抑制噪声。最后,该研究应用Faster R-CNN 模型在染色体实例图像上完成检测和分类任务。Liu 等人(2022c)同样发现低分辨率图像会使染色体更加难以区分,所以提出了SRAS-Net用于低分辨率染色体分类。该方法首先通过自注意力负反馈网络(selfattention negative feedback network,SRAFBN)对图像映射空间进行约束并选择图像中的关键信息进行重建,得到高分辨率染色体图像。然后提出的图像自适应模块(image adaptive module)将统一低分辨率和高分辨率图像以满足下游网络迁移学习要求。最后SRAS-Net使用 SMOTE(synthetic minority oversampling technique)(Chawla 等,2002)算法通过与最近邻样本插值的方式合成新样本,缓解了分类网络训练时面临的性染色体数据不平衡问题。Lin 等人(2022)认为染色体实例图像来源复杂大小不一,且大规模标注存在难度,所以提出了染色体数据扩增法(chromosome data augmentation,CDA)和图像自适应接口(image adaptive interface,IAI)帮助基 于Inception-ResNet(Szegedy 等,2016)的CIR-Net(chromosome based on inception-ResNet)模型完成染色体分类任务。基于仿射变换的CDA 方法通过旋转矩阵和平移向量实现染色体的随机旋转和平移变换。IAI 模块则是一个2 维卷积层,将根据不同尺寸的染色体实例图像选择不同的方法使其统一,再利用Inception-ResNet完成分类。
5.6 基于全局和局部特征融合的分类方法
已有方法均从染色体实例全局特征出发,极少有显式学习局部特征的方法,所以Qin 等人(2019)提出了由全局尺度网络(global-scale network,G-Net)和局部尺度网络(local-scale network,L-Net)组成的Varifocal-Net。其中G-Net负责提取染色体粗粒度全局特征并采用varifocal 机制精确的找到重点区域,然后用L-Net 提取重点区域作为细粒度局部特征。最后Varifocal-Net 融合两种特征再输入多层感知机来预测染色体的类别和极性,最后采用调度策略根据领域知识调整预测结果。这一方法利用局部细节特征弥补了粗粒度全局特征的缺点,对局部细节不同的同类染色体更鲁棒,泛化能力更强。但是因为Y染色体数据不平衡,对其辨别能力较差。Wei等人(2022)同样注意到综合考虑全局信息和局部信息的重要性,提出了三阶段的输入感知和概率预测卷积神经网络(input-aware and probabilistic prediction convolutional neural network,IAPP-CNN)。输入感知模块(input-aware module)通过注意力机制,将原始图像扩展为全局尺度图像(global image)、物体尺度图像(object image)和部分尺度图像(part image)。3 个独立的CNN 特征提取器将提取3 个尺度图像的特征,然后通过概率预测模块(probabilistic prediction)分别预测它们的类别概率分布。这些结果可以作为各个分支的置信度再通过加权的方式输出最终预测结果。
5.7 基于复杂策略的分类方法
Xiao 和Luo(2021)认为现有染色体分类需要高质量的人工分割染色体实例数据集,因此选择从细胞分裂中期图像中直接预测染色体类别,提出了基于Faster R-CNN 的染色体检测方法DeepACC,将类别预测分支改造为孪生网络结构,其中间隔分支(margin branch)引入了加性角度间隔损失(additive angular margin loss),在扩大类间距离的同时缩小类内距离。而推断分支(inference branch)则与间隔分支共享大部分参数,同时启发式地选择间隔分支中预测置信度最高的样本特征作为推断分支的最后一层分类器权重,进一步缩小了类内距离。此外还设计了组内邻接损失(group inner-adjacency loss)函数,重点惩罚组内误分类现象,进一步扩大类间距离。然而DeepACC 模型无法得到染色体实例,所以需要进一步处理。Zhang 等人(2021)提出了一种交错和多任务网络(interleaved and multi-task network)方法来完成染色体分类和染色体矫直任务。第1 阶段提出了基于HRNet(Sun等,2019)的信息交错网络来提取多分辨率的特征;在第2 阶段分辨率最高的特征图被用来定位染色体起始点、终止点和弯曲点。余下的特征图融合后则分别用来预测染色体类别和极性。而预测的染色体弯曲点将被用来实现染色体矫直。但是因为结构复杂的染色体数量较少,导致该方法对于结构复杂染色体的关键点定位能力下降。Al-Kharraz 等人(2021)则想通过集成学习的方式来提高染色体分类能力。该研究通过微调多个预先训练好的卷积神经网络模型来完成染色体分类任务,并在集成学习框架中利用平均投票对结果进行组合。
5.8 总结
综上所述,染色体分类任务在染色体自动化核型相关领域中得到了最广泛的关注。研究人员重点需要解决两个方面的问题:1)染色体图像存在的固有缺陷,包括低分辨率下不同类别染色体相似性以及数据集中性染色体不平衡问题;2)由于实验手法、条带水平等因素导致的同一类别染色体之间视觉特征差异较大。
基于简单CNN 结构的分类方法为染色体分类任务定制了专门的轻量级分类网络,并不断引入深度学习领域最新研究结论来提高性能。但基于经典CNN 结构的方法也备受关注,研究人员通过分析染色体数据特点优化创新经典分类模型。基于特征对比的分类方法则一般通过孪生网络或三元网络架构提取特征,然后利用度量学习得到判别能力较强的特征分布,最后再通过简单分类器和分类方法完成分类。基于图像预处理的分类方法则会尝试在输入分类模型前生成高分辨率图像、增强染色体条带特征、扩增染色体数据和统一染色体尺寸来提高分类性能。但是仅提取全局特征可能无法发现不同类别染色体之间的细微差异,所以基于全局和局部特征融合的方法会利用独立的分支分别提取染色体的全局特征和局部特征,再融合预测染色体类别。最后,研究人员还考虑从细胞分裂中期图像中直接完成染色体分类,或通过多分辨率特征图在完成染色体分类的基础上同时实现染色体极性预测和矫直,再或通过集成学习的方式提升分类性能,这些方法可以统称为基于复杂策略的分类方法。
展望未来,基于Transformer 的图像分类模型因其长距离建模能力而备受关注,染色体的条带模式天然具备序列形式,所以基于Transformer 的分类网络在染色体分类领域也大有可为,但是大规模高质量染色体分类数据集是此类方法成功的前提。与此同时,细胞遗传学家对于染色体形态结构特别是条带模式进行了长时间的研究,积累了宝贵的领域知识如染色体的标准模板等,所以未来也可以充分利用标准模板通过对比学习或度量学习的方式来减少数据量需求,提高分类性能。染色体分类方法的简要总结如表5所示。

表5 染色体分类方法简要总结Tabel 5 A brief summary of the chromosome classification methods
6 染色体异常
6.1 问题归纳
染色体数目异常可以通过染色体计数方法精准发现,但是染色体结构异常种类繁多,仍需要经验丰富的细胞遗传学家认真识别。已有的与染色体异常相关的任务主要包括异常发现和异常生成,接下来将分开讨论。
6.2 染色体异常发现
Yan 等人(2019)将目光对准了与慢性粒细胞白血病(CML)相关的染色体平衡易位异常t(9;22)。该异常因仅与第9 号和第22 号染色体相关,所以收集了与之相关的正常和异常数据并训练ResNet网络加以识别,实现了检测染色体特定异常的功能。Li等人(2020)尝试发现染色体结构异常中的像素级差异,因此提出了基于异常检测和染色体分类的有监督多任务学习模型CS-GANomaly(classificationenhanced GANomaly)。该方法基于cGAN架构,将异常检测网络作为判别器,而染色体分类网络将与判
别器共享底层参数,综合两个预测结果即可完成染色体异常检测任务。其中,染色体分类网络除了常规的24 类外,还将预测异常类,其中包括了所有异常染色体。如此,通过多任务训练就可以将异常检测任务转变为监督学习过程。通过联合训练,不仅学习了各类染色体正常样本的特征,还学习得到每个类别的正常样本和异常样本在高维特征空间的分布,使得模型对异常更敏感。
6.3 染色体异常生成
尽管可以从临床收集染色体异常数据,但是由于遗传性疾病较为罕见,很难构建较大的染色体结构异常数据集,所以有相关工作探索通过生成对抗网络合成染色体结构异常数据集。Uzolas 等人(2022)运用二维染色体条带分割掩码对pix2pix 图像翻译网络进行调整,使得模型能够按照用户定义的条带模式合成真实染色体和具有结构类型异常的染色体。该方法首先在染色体密度曲线上应用非线性滤波器(non-linear filter)得到染色体的条带模式和条带分割掩码,将其作为pix2pix 模型的源域,然后进行图像翻译即可得到外观真实的染色体。为了模仿异常的染色体,该研究基于Perlin 噪声随机生成Perlin 条带和异常的条带分割掩码,再通过正常样本训练好的生成器就可以合成异常染色体。
6.4 总结
综上所述,目前研究人员主要研究染色体异常发现和染色体异常生成。两类问题面临的共同难点是复杂多变的异常情况,缺失、倒位以及染色体之间的罗氏易位和平衡易位等均会产生异常染色体。针对这一问题,研究人员一方面通过缩小异常情况范围,收集专门数据并通过经典模型分类;另一方面,通过生成式对抗网络机制,共享异常检测网络和染色体分类网络,从而使得模型也能以监督学习的方法找到正常和异常染色体特征的分布规律。而为了弥补异常染色体数据的不足,研究人员也提出通过图像翻译的方式,在找到真实条带模式与真实染色体图像映射关系后,可以通过人工异常条带合成异常染色体。
染色体异常发生机制预示着针对染色体整体设计的网络可能无法有效应对染色体微结构变化。而且现有方法无法识别异常的具体类型,出具符合规范的异常诊断结果。未来研究人员可能需要尝试细化至条带级(banding level)染色体微结构特征,通过图神经网络等具有关系推理能力的神经网络发现染色体和染色体之间微结构关系变化,完成异常诊断任务。染色体异常方法的简要总结如表6所示。

表6 染色体异常方法简要总结Tabel 6 A brief summary of the chromosome anomaly methods
7 数据集
高质量标注数据是基于深度学习的染色体核型自动分析算法的性能保障。现有方法所使用的数据大多来自合作医疗机构,由于隐私限制通常无法公开获取。但仍有少量工作使用公开数据集或选择开放所使用的脱敏临床数据,现总结如下:
1)Overlapping chromosome 数据集(https:∕∕ www.kaggle.com∕datasets∕jeanpat∕overlapping-chromosomes)。如图6 所示,该数据集属于半合成重叠染色体数据集,用于染色体分割任务。该数据集将DAPI(4′,6-diamidino-2-phenylindole)染色的染色体图像和Cy3(cyanine3)荧光端粒探针图像相结合形成灰度染色体,然后两两重叠,生成13 434 幅大小为94 × 93 像素的重叠染色体图像,其中像素被标记为“背景”、“1号染色体非重叠区域”、“2 号染色体非重叠区域”和“染色体重叠区域”4个类别。

图6 Overlapping chromosome 数据集Fig.6 Overlapping chromosome dataset
2)ChromSeg 数据集(http:∕∕www.bio8.cs.hku.hk∕bibm∕)。该数据集采集了345幅大小为256 × 256像素的交叉重叠染色体图像,用于染色体分割任务。其中包括230 幅训练集图像和115 幅测试集图像。如图7 所示,专家们手动标注了“交叉重叠区域”和“染色体前景”两个类别。

图7 ChromSeg 数据集Fig.7 ChromSeg dataset
3)Clinical chromosome instance segmentation 数据集(https:∕∕github.com∕CloudDataLab∕ Overlapping-ChromosomeInstanceSegmentation)。该数据集是重叠和粘连染色体簇实例分割数据集。作者从广东省妇幼保健院采集了1 655个脱敏染色体簇图像,总计4 766 个染色体实例,由专家利用LabelMe 工具箱进行实例分割标注。数据集以8∶1∶1 的比例被划分为训练集(1 324)、验证集(165)和测试集(166)。
4)Chromosome cluster types identification数据集(https:∕∕github.com∕ChengchuangLin∕Chromosome-ClusterIdentification)。该数据集从广东省妇幼保健院收集了6 592个大小为224 × 224像素的脱敏染色体簇图像,用于染色体簇分类任务。如图8 所示,根据染色体实例之间的交集和连通性,人工标注依次为“实例”、“粘连”、“重叠”和“粘连—重叠”4种类型。

图8 Chromosome cluster types identification 数据集Fig.8 Chromosome cluster types identification dataset
5)BioImLab classification 数据集(http:∕∕bioimlab.dei.unipd.it∕Chromosome%20Data%20Set% 204Class.htm)。如图9 所示,该数据集由专业的细胞遗传学家从119个正常和异常细胞中人工分割得到5 474幅单条Q显带染色体图像并进行分类标注。所有染色体均根据ISCN规则进行旋转使其处于极化状态。

图9 BioImLab classification 数据集,2号染色体Fig.9 BioImLab classification dataset,chromosome 2
6)Chromosome-images 数据集(https:∕∕github.com∕Xi-Hu∕Chromosome-Images)。该数据集属于染色体分类数据集,利用主动轮廓模型(active contour model)从91 个核型图中分割得到4 184 幅G 显带单条染色体图像,并通过扩展背景统一大小为100 ×220 像素。其中9 号和20 号染色体181 条,X 染色体152条,Y染色体30条,其余各号染色体均为182条。如图10为3号染色体示例。

图10 Chromosome-images数据集,3号染色体Fig.10 Chromosome-images dataset,chromosome 3
7)CIR-Net 数据集(https:∕∕github.com∕Cloud-DataLab∕CIR-Net)。该数据集从广东省妇幼保健院收集了32 个男性和33 个女性脱敏核型图并人工分离出总计2 990 幅G 显带单条染色体图像用于评估染色体分类模型性能。其中1—22 号染色体各有130幅,X染色体总计98幅,Y染色体32幅。图11为CIR-Net数据集4号染色体示例。

图11 CIR-Net数据集,4号染色体Fig.11 CIR-Net dataset,chromosome 4
以上整理了7 个公开可获取数据集,分布在染色体分割、染色体簇分类和染色体实例分类领域。可以发现,染色体相关数据集规模均不大,难以支撑复杂模型取得较好效果。其次,现在尚无针对其他分析任务的公开可获取数据集。综上所述,应该广泛支持和激励研究人员大规模收集并高质量标注各个染色体分析任务所需数据,经过脱敏处理后使其公开可获取,以支持染色体核型自动分析算法快速向前发展。
8 讨论
基于深度学习的染色体核型自动化分析方法借助其数据驱动特点和强大的学习能力在许多复杂的染色体核型分析任务上都取得了长足进步。本文收集了大量基于深度学习的染色体核型自动分析方法,分别面向染色体计数任务、染色体分割任务、染色体簇分类任务、染色体预处理任务、染色体分类任务和染色体异常分析任务。尽管这些方法针对染色体任务和数据特点进行了性能优化,但仍存在一些不足。本文分析上述研究工作趋势,并提出了未来发展方向。
8.1 研究趋势
针对染色体计数任务,已有方法主要通过目标检测框架解决,而需要解决的问题也主要是自相似性问题和染色体粘连重叠带来的错误去冗余和定位不精确等问题。另外也有研究工作思考如何加速染色体计数模型推理速度,使其更贴近临床场景。
针对染色体分割任务,已有方法大致可以分为语义分割模型和实例分割模型,但前者仅能解决两条或多条染色体重叠形成的染色体簇的分割问题,后者虽然基本能实现自动化染色体分割,但仍需要标注关键点等额外监督信息。
针对染色体簇分类任务,已有方法大致会根据不同的标准将染色体簇进行划分,一种是根据重叠染色体的数目进行划分,另一种则是根据粘连和重叠染色体的相互关系进行划分。但是从模型角度观察,目前研究工作乏善可陈,仍有很大的创新空间。
针对染色体预处理任务,已有方法主要解决细胞分裂中期图像降噪和染色体矫直两项预处理任务。其中细胞分裂中期图像降噪任务被转化为分割任务,需要将染色体所属区域与背景和图像中存在的杂质分开。而已有的染色体矫直方法均依赖于生成式对抗网络通过图像翻译或运动变换将弯曲染色体矫直。但是生成的染色体能否被用于后续核型分析和临床诊断仍需更多临床实验证明。
针对染色体分类任务,得益于基于深度学习的图像分类网络的蓬勃发展,染色体分类相关工作也在核型分析相关任务中得到了最多的关注和发展。已有方法不管是简单的CNN 方法还是采用复杂策略加以解决,都需要针对性地解决染色体分类任务中存在的数据量不足、低且不一致的分辨率和同类染色体形态差异较大但不同类别染色体相似程度高等问题。
针对染色体异常分析任务,虽然临床专家高度关注,但目前提出的工作仅能通过CNN 检测特定的染色体异常,或通过生成式对抗网络框架简单判断是否存在异常,以及通过神经网络人工合成异常染色体。其主要原因在于染色体结构异常复杂多变,且难以大规模收集并标注异常数据集,因此还需要持续关注和进一步研究。
8.2 未来发展方向
本文在总结分析每个染色体核型分析任务相关工作的基础上,针对尚需继续研究的难点挑战给出了许多具体可行的研究方向。接下来将从染色体核型分析整体出发,给出该领域未来发展方向。
1)目前基于深度学习的染色体核型分析工作主要集中在染色体分割和染色体分类两个任务上,这与深度学习在自然图像领域的发展趋势一致。但是,诸如染色体计数、染色体簇分类、染色体预处理以及染色体异常分析等任务由于没有清晰明确的问题定义,也缺乏公开可获取的数据,导致这些重要任务尚未被研究人员广泛关注。所以通过本文归纳整理的相关工作和数据,有希望引起更多研究人员关注。
2)本文认为至关重要的核型诊断任务目前尚未引起关注。核型诊断任务需要从核型图中发现明确的异常并给出符合规范的诊断结果。因为染色体结构异常种类纷繁复杂,因此无法简单地从染色体实例级别(instance level)的分析入手,而需要细化至染色体微结构,即条带级别(banding level)分析。为了得到符合规范的诊断结果,还需要结合领域知识通过图神经网络等具有推理能力的方法,分析染色体微结构之间的复杂关系(如易位、倒位等)。
3)众所周知,深度学习方法需要大规模高质量的标注数据,但完成染色体数据标注需要经验丰富的医学专家,限制了数据标注规模。而现在广泛应用的大规模自然图像数据集(如ImageNet等)与染色体也存在明显的语义鸿沟。现阶段染色体核型分析过程中将人工产生许多中间分析结果,同时还有长期积累的领域知识作为辅助监督信号。所以,可以考虑采用半监督学习(semi-supervised learning)、弱监督学习(weakly-supervised learning)和自监督学习(self-supervised learning)方法,在大量无标注或弱标注数据中学习染色体特征分布,再通过小规模高质量标注数据微调来适配各个下游任务,避免过高的标注成本。
4)人工合成数据是应对数据匮乏的重要手段,在染色体核型分析领域更可以发挥重要作用。从方法角度,生成式对抗网络和扩散模型等现阶段广泛应用的生成模型都可以用来进一步探索染色体数据合成。从应用角度,生成模型可以在矫直弯曲染色体和合成结构异常染色体方面发挥重要作用。但值得注意的是,染色体图像合成不同于自然图像合成,错误合成数据有可能导致严重医疗事故,造成严重后果。因此与临床专家合作设计流程清晰完备的验证方法也是未来需要重点关注的方向。
5)基于多模态医学数据的分析方法在医学影像分析领域被广泛应用。在多模态染色体核型分析领域,可以选择不同成像原理的图像,例如G显带染色体和荧光原位杂交染色体图像相结合;也可以选择不同模态数据,例如染色体图像和核型诊断报告相结合;更可以选择不同实验数据,例如染色体图像和基因测序数据相结合等。通过不同模态数据组合,可以进一步拓展染色体核型分析应用边界,更好地满足临床需求。
9 结语
本文对基于深度学习的染色体核型自动化分析方法进行了比较全面的综述。染色体核型分析应用前景广阔,需求日益上涨,因此染色体核型自动化分析方法成为研究热点。但是传统的自动化方法在面对形态结构复杂、成像质量波动较大的染色体目标时,性能和泛化能力都不足以应对复杂的临床需求。因此具有强大学习能力和数据驱动特点的深度学习方法成为实现染色体核型自动化分析的新热点,性能和泛化能力都有了较大进步。本文系统总结了染色体计数、染色体分割、染色体簇分类、染色体预处理、染色体分类和染色体异常分析等6 大染色体核型自动化分析任务。具体来说归纳了任务解决框架,阐述了已有解决方案,说明了还未解决的难点挑战并最终为每项任务提出了具体可行的发展方向。数据是深度学习方法的性能保障,但大多数染色体核型分析相关工作所使用的数据主要来源于合作医疗机构。本文还收集整理了7 个公开可获取的染色体核型分析相关数据集,供研究人员进一步开发高性能算法。最后,从染色体核型自动化分析整体出发,提出了5个未来可能的发展方向。
综上所述,基于深度学习的染色体核型分析方法逐渐成为自动化核型分析的主流。但该领域与临床紧密结合,不仅需要继续在模型方法上研究探索,还需要与医学专家紧密协作。通过在临床实践中广泛应用和评价,研究人员可以从中发现新的问题加以抽象并提出新的解决方案。所以,基于深度学习的染色体核型自动化分析方法仍有极大的发展空间。
猜你喜欢
宁夏医学杂志(2020年3期)2021-01-21
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
哈尔滨医药(2015年2期)2015-12-01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
中国当代医药(2015年9期)2015-03-01
应用海洋学学报(2014年3期)2014-11-22
高中生学习·高三版(2014年3期)2014-04-29
高中生学习·高三版(2014年3期)2014-04-29
