
婴儿左颞骨朗格汉斯细胞组织细胞增生症1例报告并文献分析
2023-11-12 02:29张贤华樊金星邱文琪
南昌大学学报(医学版) 2023年5期
张贤华,樊金星,邱文琪,杨 明
(江西省儿童医院神经外科,南昌 330006)
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)是一种罕见的全身性疾病,其特点是来自骨髓髓系祖细胞的朗格汉斯细胞的增殖和积累,病因和发病机制不明[1]。LCH好发于儿童,发病率约为4~8例/百万人,成人LCH很罕见,发病率约为1~2例/百万人[2]。目前,国内外文献对累及婴儿颞骨乳突部的LCH鲜见相关报道,江西省儿童医院神经外科2021年3月收治1例,现就其临床特点、诊疗经过进行回顾性分析,为临床诊治提供参考。
1 临床资料
1.1 一般资料
患儿,男,7个月零5 d,因“发现左耳后肿物2周”入院,肿物呈渐进性增大,无破溃,无任何鼻咽症状。入院查体:左耳后可触及一大小约4.0 cm×3.5 cm×3.5 cm肿物,质硬,无触痛,边界不清,活动度差,左侧外耳道无分泌物,耳膜完整。患儿监护人知情同意并签署手术知情同意书。
1.2 影像学及听力检查结果
头颅CT,提示左侧颞骨乳突部骨质呈溶骨吸收性破坏,相应区可见较大肿块影,最大截面大小约3.5 cm×3.5 cm×4.0 cm,向颅内侵犯(图1)。头颅MRI,提示左侧颞骨可见团状异常信号,T1等低信号,T2稍高信号,信号欠均匀;FLAIR呈不均匀混杂信号;注药后呈明显强化,强化不均匀;包块突向颅内,紧贴小脑边缘(图2)。头颅MRA提示左侧乙状窦显示不清。双侧内耳MRI提示双侧耳蜗、前庭、半规管、内听道形态可,未见明显异常信号,双侧耳蜗神经及前庭神经分别入耳蜗及前庭,双侧中耳腔未见明显异常信号。患儿听性脑干反应检查骨导反应阈正常,气导反应阈升高至50~60 dBHL,表现为传导性听力的损失。

A:左侧颞骨乳突部相应区可见较大肿块影,最大截面大小约3.5 cm×3.5 cm×4.0 cm,向颅内侵犯;B:左侧颞骨乳突部骨质呈溶骨吸收性破坏。

A—B:平扫,左侧颞骨可见团状异常信号,T1等低信号,T2稍高信号,信号欠均匀;C—D:增强,注药后呈明显强化、强化不均匀,包块突向颅内、紧贴小脑边缘;E—F:平扫(冠状位),左侧颞骨可见团状异常信号,T1等低信号,T2稍高信号。
1.3 诊治经过
术前结合患者病史、影像学检查初步诊断为左颞骨肿瘤。拟在面神经监测下行左颞骨肿瘤切除术。
手术过程:取左侧耳后弧形切口,见皮下瘤体呈淡红色鱼肉状,边界不清,血供丰富,肿瘤向乳突骨侵蚀,深入到乙状窦硬膜,乙状窦受压闭塞,部分侵蚀外侧及后半规管骨,上及中颅窝硬膜及后管壁。显微镜下分块切除肿瘤,用双极烧灼术切除中、后颅窝与硬脑膜关系密切的肿瘤,全切肿瘤。
1.4 治疗结果
1)手术结果及并发症:患儿获得肿瘤全切除。
术后患儿出现口角歪斜,完善面神经传导速度检查提示面神经损伤可能,予以康复治疗后好转。未发生颅内感染等其他并发症。
2)术后病理学检查结果:HE染色显示朗格汉斯组织细胞,背景中可见淋巴细胞、中性粒细胞、嗜酸性粒细胞浸润,周边见少量多核细胞(图3A—B)。免疫组织化学染色显示朗格汉斯组织细胞增多症,CD1a(+)、Langerin(+),S-100(+),BRAFV600E(+)(图3C—F)。
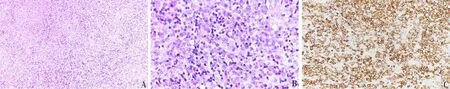
A:低倍镜下,显示朗格汉斯组织细胞(HE×100);B:高倍镜下,显示朗格汉斯组织细胞,背景中可见淋巴细胞、中性粒细胞、嗜酸性粒细胞浸润,周边见少量多核细胞(HE×400);C:免疫组织化学染色,提示CD1a(+)(Envision×200)。

D:免疫组织化学染色,提示Langerin(+)(Envision×200);E:免疫组织化学染色,提示S-100(+)(Envision×200);F:免疫组织化学染色,提示BRAFV600E(+)(Envision×200)。
3)术后治疗及随访结果:术后完善肺部、脊柱、四肢、骨盆X线及肝脾彩超等相关检查均阴性。术后转血液内科行化疗,应用长春新碱(VCR)+泼尼松(PDN)方案。第1—6周:第1—4周,PDN 40 mg·m-2·d-1,第5—6周减停;VCR 1.5 mg·m-2,每周1次。第7—26周:PDN 40 mg·m-2·d-1,每3周的第1—5天;VCR 1.5 mg·m-2,每3周1次。
随访1年,患儿肿瘤无复发,未出现新损害。
2 讨论
LCH的病因目前尚不明确,有研究[3-6]认为可能与免疫功能紊乱、细胞因子介导、病毒感染和遗传等因素有关。LCH可累及骨、皮、脑下垂体、肺、中耳、颈淋巴结、肝、脾、神经系统等全身任何器官。骨骼是最常受累及的系统,大约80%的LCH患者存在骨病损,并且其中一半的病变是单一的[7-8]。最常见的骨受累部位是颅骨,超过2/3的骨病患者会累及颅骨,其次是脊柱、四肢和骨盆[9]。颅底骨通常受累,颞部颅骨突出或中耳炎或外耳炎是常见的症状。因此,累及颞骨乳突部时容易造成延误诊断或误诊。所以,有必要对婴儿颞部的LCH的临床表现、影像学特点、治疗方法及预后情况加以总结,以期提高对婴儿颞部LCH的认识及治疗效果。
2.1 临床表现
LCH的临床表现与其受累的部位密切相关,根据病变累及器官或系统的数量分为单系统LCH和多系统LCH,单系统LCH指仅累及单个器官或系统(如皮肤、骨骼、淋巴结等),多系统LCH指累及2个或2个以上的器官或系统。根据是否累及危险器官或系统分为低风险和高风险组,高风险组累及的器官或系统主要包括造血系统、肝脏、脾脏[10]。病变侵犯颞部时主要表现为颞部包块、听力异常等。颞部LCH患儿的听力下降多数是因为病变堵塞外耳道及鼓室引起。IRVING等[11]对131例LCH患者(平均年龄3.2岁)进行了一项回顾性分析,发现73%的病例累及头颈部;颞骨受累占其中的19%,表现为乳突肿胀、耳息肉、耳漏、耳聋,偶见耳痛;耳息肉的特征性表现为起源于乳突,侵蚀后上管壁,鼓膜基本正常,有助于与急性乳突炎鉴别。TOS[12]回顾性分析500例LCH患者资料,发现面瘫发生率为2.8%。
2.2 影像学特点
影像学检查是目前颞骨LCH的主要检查手段。颞骨LCH的CT多表现为颞部软组织肿块影,密度大多不均匀,周围骨质呈溶骨性破坏,仅边缘可残留少许骨质,轮廓不规则,边缘欠清晰[13]。CT与MRI相比,能够清晰显示颞骨的骨质破坏程度,确定骨质破坏的范围,显示病变与周围组织器官的关系。头颅MRI在显示骨质破坏的程度、范围方面虽然不如CT,但在显示病变的性质和范围,与周围组织器官的关系等方面强过CT。
2.3 诊断与鉴别诊断
LCH是一种非常罕见的疾病,由于不同的临床和影像学表现,临床上很难诊断,诊断的金标准是病理组织活检。LCH的主要病理变化为病理组织中存在朗格罕斯细胞(Langerhans cell,LC)的异常增生,淋巴细胞、中性粒细胞、嗜酸性粒细胞的浸润。确诊的主要免疫组织化学检查包括巨细胞的Langerin(CD207)、S-100、CD1a等阳性表达,并且可以在细胞质中发现Birbeck颗粒[14]。颞骨LCH常易与耳部肿瘤性等疾病相混淆,易造成误诊和漏诊。颞骨受累时要注意与横纹肌肉瘤相鉴别,影像学上,仅累及颞骨前部尤其是岩顶和中耳的病变更容易发生横纹肌肉瘤,累及颞骨后部(乳突)的病变更容易发生LCH[15]。
2.4 治疗方法
由于LCH属于临床少见病,目前尚无标准的治疗方法,治疗要根据病情的轻重和程度。治疗策略包括单纯观察、手术切除、口服类固醇药物、放射治疗和化疗等[16-17]。对于局部皮肤病变的低危型患者通常观察或使用局部类固醇治疗,当疾病非常广泛或已被证明对局部难以治疗时,可能需要全身治疗。对于骨骼受累的单系统患者,目前通常采用手术清除病灶,术后联合化疗的治疗方案[18];多发及多系统受累可行全身化疗;因有发育延迟和畸形、继发肿瘤等相关后遗症,目前儿童一般不再推荐放疗[19]。因本例患者为颅骨受累的单系统患儿,所以采用了手术切除,术后联合化疗的治疗方案[18]。同时因位于颞骨的LCH因常累及面神经,侵犯硬脑膜、乙状窦等,术中应注意保护。本例患儿术后出现口角歪斜,考虑术中牵拉可能。
2.5 预后
LCH的预后主要与患儿发病的年龄、组织器官受累的范围、有无功能受损以及受损的程度、治疗等有密切的关系。研究发现,年龄较小(<2岁)的多系统LCH伴器官功能障碍是预后不良的指标,而单系统LCH预后较好[4]。有研究[19]表明,多系统LCH患儿1、3及5年的总生存率分别为79%、74%和71%,累及肝脾等高危器官的LCH患者生存率更低,1年生存率仅为33%,5年生存率仅为25%。因此,对于多系统性LCH,尤其是累及肝脾等高危器官的患儿[16],病情十分凶险,进展迅速,需要进行积极的治疗。
综上所述,婴儿颞部LCH临床上少见,目前尚无统一的治疗策略。外科手术切除仍是当前的主要治疗手段。对于单发的颞骨LCH,手术切除过程中应注意面神经的保护,因有发生面神经损害的风险。然而,这种风险可以通过术中使用面神经监测和全面的解剖学知识避免。同时需注意,若病灶侵犯硬脑膜、横窦、乙状窦等时,在行病灶切除时要注意保护,切勿损伤。
猜你喜欢
国际放射医学核医学杂志(2020年2期)2020-05-30
中国美容医学(2019年9期)2019-09-20
中国美容医学(2017年6期)2018-02-05
中华耳科学杂志(2017年4期)2017-09-18
临床医药文献杂志(电子版)(2017年10期)2017-05-18
临床医药文献杂志(电子版)(2017年16期)2017-03-07
反射疗法与康复医学(2017年7期)2017-01-16
中国美容整形外科杂志(2015年1期)2015-08-27
教育与职业(2014年28期)2014-01-19
中国实用医药(2013年32期)2013-09-16
