
二黄栓制备工艺及其含量测定方法*
2023-11-06 15:58:18苟欢刘嵘熊微施春阳方建国王文清
医药导报 2023年11期
苟欢,刘嵘,熊微,施春阳,方建国,王文清
(华中科技大学同济医学院附属同济医院 1.药学部;2.妇科,武汉 430030)
二黄栓由雄黄、黄连等药味组成,临床用于治疗高危型人乳头瘤病毒持续感染的子宫颈炎症及子宫颈上皮内瘤样病变Ⅰ级[1]。笔者在本研究采用适宜的制剂技术开发其栓剂,按中药新药分类本品为在研1.1类新药。通过热融法使药物以无定形态均匀分散在基质中,形成栓剂,在腔道经黏膜吸收,可规避药物首关效应,维持血药浓度稳定,获得更优药效。通过考察不同基质类型和制备条件对制备工艺的影响,经G1-熵权法结合正交实验设计建立二黄栓的制备工艺,并利用雄黄与其他原辅料理化性质差异考察不同前处理方法对二黄栓中二硫化二砷(As2S2)含量测定的影响,建立了碘量法和原子吸收分光光度法(atomic absorption spectrometry,AAS)。
1 仪器与试药
1.1仪器 AA-6300C原子吸收分光光度仪(日本岛津公司);HL-1型砷空心阴极灯(河北宁强光源有限公司);HeraeusMultifuge X1R离心机(赛默飞世尔科技公司);AUW220D分析天平(日本岛津公司,感量:0.01 mg);RB-1融变时限测试仪(天津市国铭医药设备有限公司);JHH-6数显恒温水浴锅(金坛市精达仪器制造有限公司)。
1.2试药 砷元素标准溶液(国家有色金属及电子材料分析测试中心,批号:223004-6,1 000 μg·mL-1);二黄栓(自制,批号:20220212,20220314,20220510,20221109,20221221),水飞雄黄粉(湖南柳城中药饮片有限公司,批号:210105),其余试剂均为分析纯。
2 方法与结果
2.1评价指标赋权
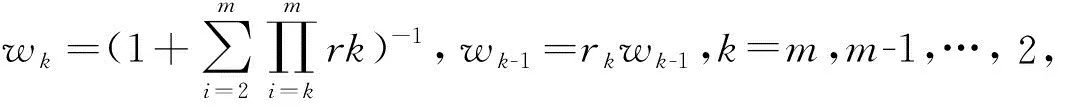
2.1.2熵权法 熵权法是根据各项指标值的离散程度确定权重,避免了人为因素带来的偏差,能更好地解释所得结果,计算过程参考柯寄明等[4-5]所用方法,得客观权重w2。
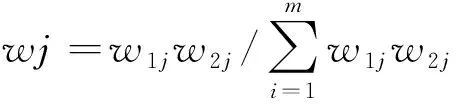
2.2二黄栓的制备工艺
2.2.1二黄栓的基质处方优化 由于基质类型及用量对栓剂的成型工艺具有较大影响,因此考察了36型混合脂肪酸甘油酯(油脂性)、硬脂酸聚烃氧(40)酯(水油兼容性)、甘油明胶(水溶性)3种类型及其用量(每粒0.66、0.99、1.32 g)对二黄栓成型工艺的影响。通过设计L9(34)正交实验,采用 G1-熵权法对外观性状、质量差异和融变时限进行综合评分,按“2.1节”下计算得各评价指标的权重值,见表1。
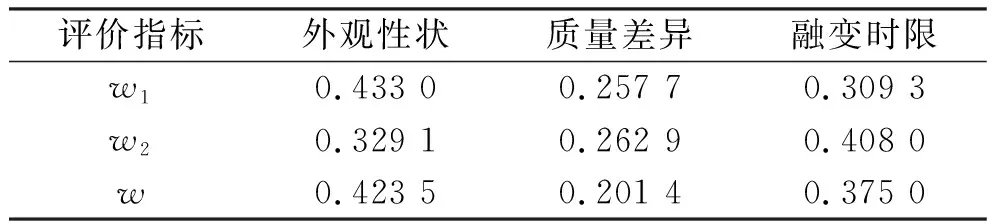
表1 基质处方优化评价指标的权重值Tab.1 Weight values of matrix prescription optimization evaluation indexes
由表2可知,基质类型对栓剂制备工艺的影响程度大于基质用量,其中A1>A2>A3,B2>B1>B3,根据表3可得,仅基质类型对成型工艺有显著影响,结合直观和方差分析结果,选用最佳基质为混合脂肪酸甘油酯,用量为每粒0.99 g,与药味总量配比为3:1。
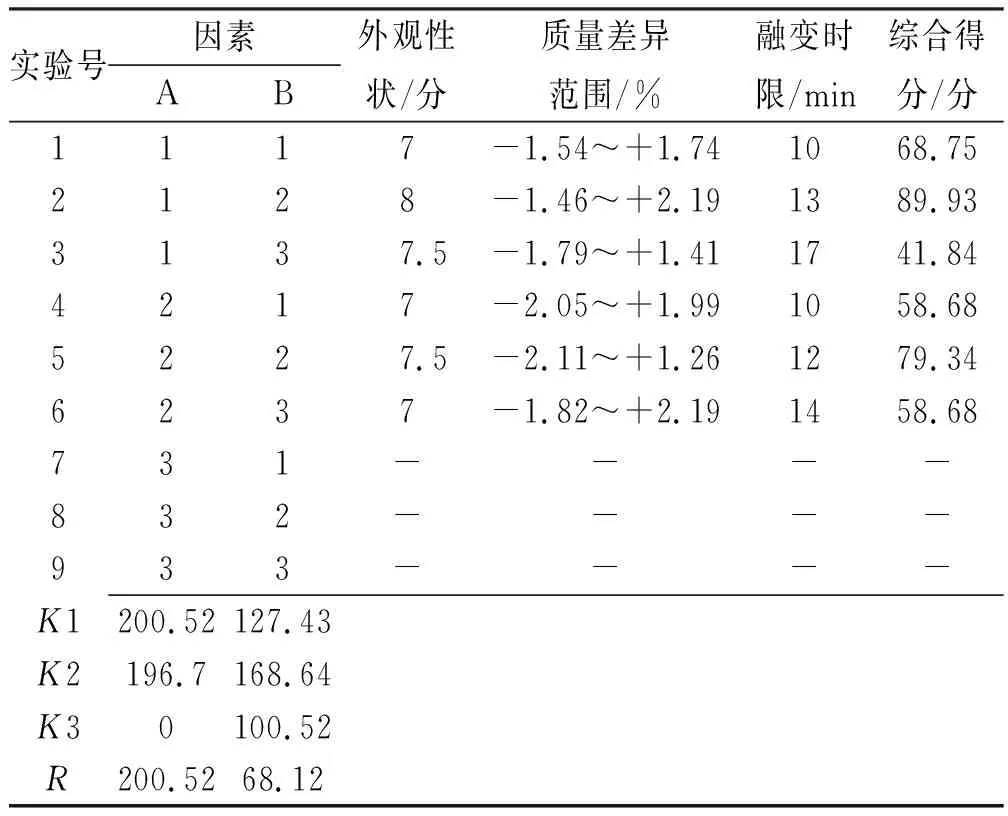
表2 二黄栓基质处方优化L9(34)正交实验结果Tab.2 L9(34)orthogonal results of matrix formulation optimization for Erhuang suppository
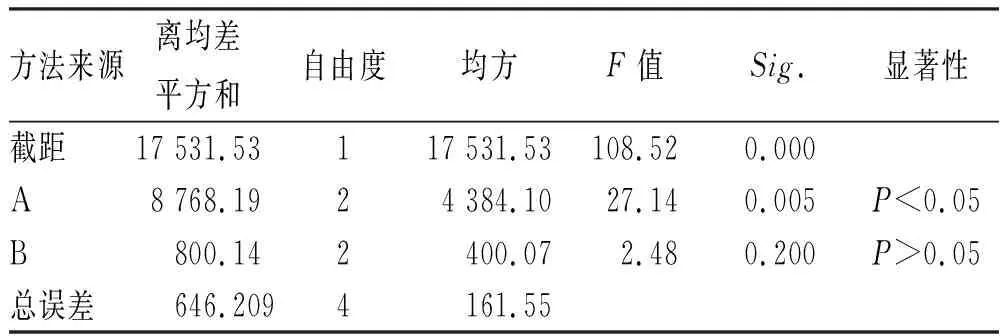
表3 基质处方优化方差分析表Tab.3 Results of variance analysis of matrix formulation optimization
2.2.2二黄栓的制备条件优化 考察基质熔融温度、搅拌时间、注模温度对制备工艺的影响,通过设计L9(34)正交实验,采用G1-熵权法对外观性状、质量差异和融变时限进行综合评分,结果见表4—6。
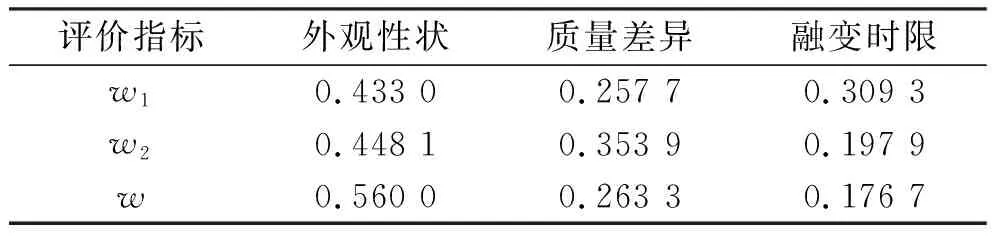
表4 制备工艺评价指标的权重值Tab.4 Weight values of preparation process evaluation indexes
由表5可知,各因素对栓剂制备工艺影响程度:基质熔融温度>搅拌时间>注模温度,其中A2>A1>A3,B2>B3>B1,C3>C1>C2;由表6可得,3个因素对制备工艺均无显著影响。结合直观和方差分析结果,最终优选制备条件为A2B2C3。即基质置60 ℃水浴至熔融状态,再按处方比例加入各药粉搅拌20 min至溶液均匀,50 ℃灌注,冷却成型,即得。
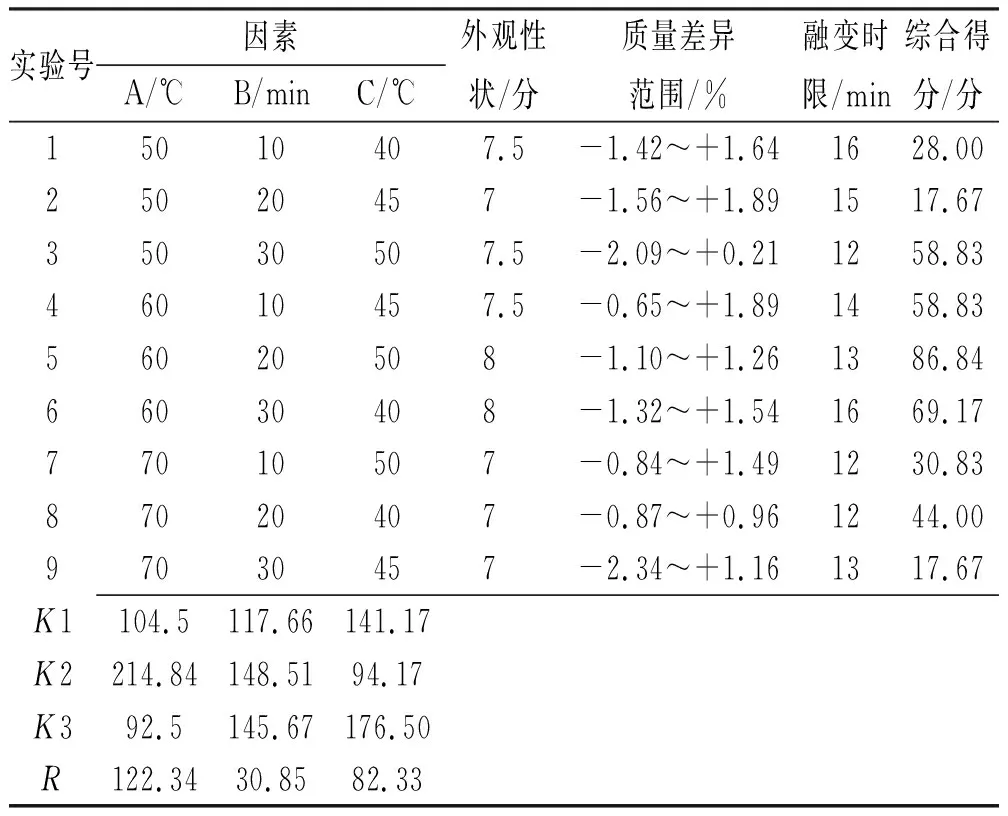
表5 二黄栓制备工艺的L9(34)正交实验结果Tab.5 L9(34)orthogonal results of preparation process for Erhuang suppository
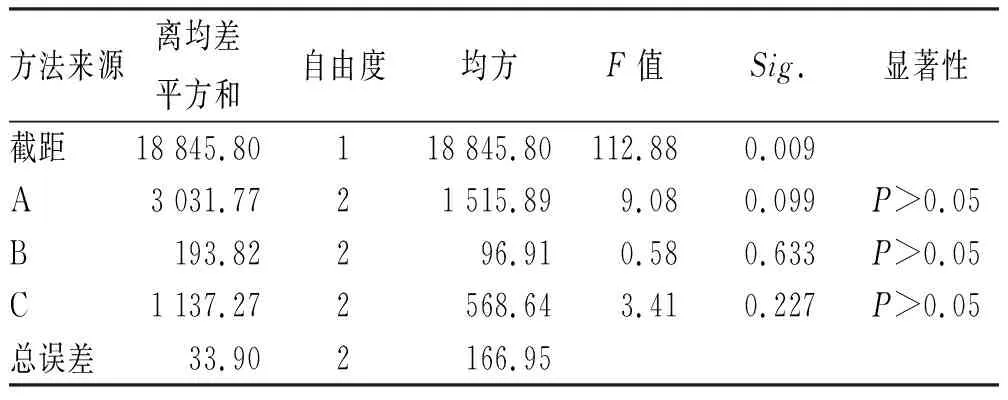
表6 制备工艺的方差分析Tab.6 Results of variance analysis of preparation process
2.3工艺验证 根据最佳制备工艺,按处方比例称取并制备二黄栓3批。最终栓剂的各指标等均符合《中华人民共和国药典》2020年版规定,表明该制备工艺稳定、重现性好,可进行大规模生产。
2.4不同前处理方法测定二黄栓中As2S2的含量 碘量法测定中成药中矿物药时,取样量、试剂用量及滴定液用量等因素直接影响滴定结果的准确性。通过高效的前处理方法除去基质等物质后可减小消解试剂用量,减小干扰,并增大滴定液用量,减小误差,是消解法与碘量法结合测定的关键点。根据雄黄和其他原辅料的理化性质差异,选择下列方法进行考察,按“2.5.2”和“2.6.3”方法测定含量,结果见表7。

表7 不同前处理方法对二黄栓含量测定的影响Tab.7 Effect of different pretreatment methods on content determination of Erhuang suppository
由表7可知,碘量法测定时,直接消解处理耗时长效率低。因雄黄粉易吸附管壁,经离心和过滤处理后含量偏低,以上3种方法不适于本栓制备。结果表明经水和正丁醇除杂富集后进行消解为碘量法最佳前处理方法,该方法进行优化后最终条件为水和正丁醇的比例为1:3、水浴温度为80 ℃、消解液用量为16 mL。AAS测定时,采用直接消解法和萃取-消解法测得As2S2含量较高,前者取样量减少时,消解时间可缩短,步骤简单,操作方便,因此选择直接消解。
2.5碘量法测定As2S2含量方法学考察
2.5.1对照品贮备液 取雄黄对照品(本课题组精制,经碘量法测定As2S2含量为99.38%)约1.25 g,精密称定,置凯式定氮瓶,按“2.5.2节”方法,定容至250 mL,得As2S2含量为5.00 mg·mL-1对照品贮备液。
2.5.2供试品溶液 取二黄栓5粒,切碎,混匀,取约1.5 g,精密称定,置锥形瓶,加正丁醇30 mL和水10 mL,80 ℃水浴加热至全部融化,振摇,静置,趁热除去正丁醇层液体,蒸干水层液,加硫酸钾2 g、硫酸铵5 g与硫酸20 mL,后续照《中华人民共和国药典》(2020年版)一部雄黄含量测定[6]项下进行,即得供试品溶液。采用碘滴定液进行滴定,每毫升碘滴定液(0.05 mol·L-1)相当于As2S25.348 mg[6]。
2.5.3专属性考察 取处方中除雄黄外的其他药味制备阴性样品,按“2.5.2节”方法测定,结果碘滴定液消耗2滴,表明样品中其他药材对测定无干扰。
2.5.4线性关系考察 分别精密吸取对照品贮备液12、18、24、30、36、42、50 mL,加水稀释至100 mL,测定As2S2含量。以As2S2取样量为横坐标(X,mg),以碘滴定液(0.05 mol·L-1)消耗体积为纵坐标(Y,mL)。结果表明As2S2取样量在59.88~249.48 mg范围内与碘滴定液消耗体积(mL)呈良好的线性关系,回归方程Y=0.187 0X+0.527 5,r=0.999 0。
2.5.5稳定性实验 取二黄栓约1.5 g,精密称定,按“2.5.2节”方法处理并于0、2、4、8、12 h测定As2S2含量,结果RSD为2.32%(n=5),表明供试品溶液在12 h内稳定。
2.5.6重复性实验 取二黄栓约1.5 g,精密称定,共6份,按“2.5.2节”方法测定含量,结果As2S2平均含量为65.58 mg·g-1,RSD为1.26%,表明该方法的重复性良好。
2.5.7加样回收率实验 取已知As2S2含量的二黄栓约0.75 g,精密称定,共6份,精密加入雄黄对照品60 mg,按“2.5.2节”方法处理后测定含量,结果平均加样回收率为101.48%,RSD为1.16%(n=6)。
2.6AAS测定As2S2含量方法学考察
2.6.1仪器条件 采用火焰原子化器,氘灯扣除背景,以乙炔为燃气、空气为助燃气进行测定。元素:As,波长193.7 nm,灯电流10 mA,狭缝0.7 nm,燃气流量2.2 L·min-1,燃烧头高度5 mm,雾化器提升时间3 s,积分时间5 s。
2.6.2标准溶液 精密吸取1 000 μg·mL-1砷标准溶液2、4、6、8、10 mL,分置100 mL量瓶,用空白溶液(2%硝酸溶液)稀释至刻度,摇匀,即得。
2.6.3供试品溶液 取二黄栓5粒,切碎,混匀,取约0.25 g,精密称定,置锥形瓶,加硫酸钾1 g、硫酸铵2 g与硫酸8 mL,消解,放冷,置200 mL量瓶中并用水稀释至刻度,摇匀,即得。
2.6.4专属性考察 取空白溶液和阴性样品溶液,按“2.6.1节”条件测定吸光度,结果吸光度为供试品溶液的1.50%,表明其对测定无干扰。
2.6.5线性关系考察 取浓度分别为20、40、60、80、100 μg·mL-1的砷标准溶液,按“2.6.1节”条件测定吸光度,以砷元素浓度(X,μg·mL-1)为横坐标,以吸光度(Y)为纵坐标绘制标准曲线,回归方程为:Y=6.353 0×10-3X+0.055 8,r=0.998 6。结果表明砷元素的浓度在20~100 μg·mL-1范围内与吸光度的线性关系良好。
2.6.6仪器精密度实验 取砷标准溶液(60 μg·mL-1),按“2.6.1节”条件连续测定6次,结果RSD为3.17%(n=6),表明仪器精密度良好。
2.6.7稳定性实验 取二黄栓约0.25 g,精密称定,按“2.6.3节”方法处理并分别于0、2、4、8、12 h测定吸光度,结果RSD为3.66%(n=5),表明供试品溶液在12 h内稳定。
2.6.8重复性实验 取二黄栓约0.25 g,精密称定,共6份,按“2.6.3节”方法处理后测定吸光度,结果As2S2平均含量为66.09 mg·g-1,RSD为2.91%(n=6),表明该方法重复性良好。
2.6.9加样回收率实验 取二黄栓约0.125 g,精密称定,共9份,分别精密加入砷标准溶液4.8、6.0、7.2 mL(相当于样品中砷含量的80%、100%、120%),各3份,按“2.6.3节”方法处理后测定吸光度,结果平均加样回收率为99.31%~105.20%,RSD<3.05%(n=9)。
2.7样品测定结果的比较 取样品5批,分别采用碘量法和AAS测定,AAS测定的砷含量通过As与As2S2的分子量转换计算得As2S2含量。对两种测定方法结果进行t检验,结果表明差异无统计学意义,见表8。
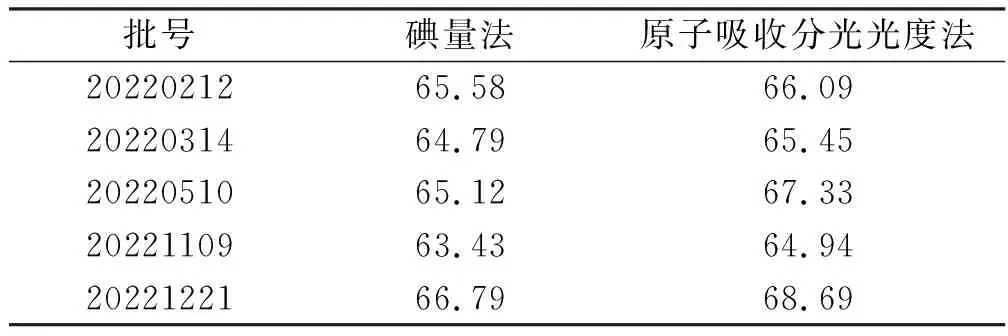
表8 5批样品测定结果Tab.8 Determination results of five batches of samples n=2,mg·g-1
3 讨论
3.1指标赋权与评价方法 在多指标综合评价中赋权方法和各指标权重最为重要。目前栓剂多采用主观赋权评价制备工艺,根据评价指标重要程度主观给予权重,可能造成权重分配不合理或与实际差异过大,最终结果缺乏准确性。通过G1法主观赋权快速计算,无需一致性检验,再结合熵权法进行客观赋权,可靠性和准确性更强[4]。本研究采用G1-熵权法组合赋权方法对栓剂制备工艺进行评价,不局限于主观评价,可同时兼顾决策者的主观性和待评价各指标的客观联系,使二黄栓制备工艺的综合评价结果更加科学准确。
3.2不同基质材料对二黄栓制备工艺的影响 栓剂基质有油脂性基质包括天然的脂肪酸和合成脂肪酸酯等,水溶性基质包括聚氧乙烯、甘油明胶、泊洛沙姆和聚乙二醇类等[7]。本研究选择几种类型中具有代表性的常用基质进行考察。在筛选基质时发现水溶性基质甘油明胶不适于本栓的制备,处方中的白矾遇水会产生氢氧化铝吸附胶体粒子,导致整体凝固,无法灌装。因此在考察基质时含白矾等栓剂应注意药物与水溶性基质是否会发生反应。水油兼容性基质硬脂酸聚烃氧(40)酯熔化时产生小气泡,在栓剂中形成气孔,造成质量差异较大。最终优选混合脂肪酸甘油酯为基质,用量为每粒0.99 g,与药味总量配比为3:1。
3.3不同制备条件对二黄栓制备工艺的影响 基质熔融温度、搅拌时间等诸多因素影响栓剂的制备工艺。基质熔融温度影响溶液流动性和混合均匀度等,熔融温度过低,溶液流动性低,药粉不易分散,造成有效成分不均匀;熔融温度过高,与注模温度温差过大,冷却时间太长,影响制备效率。搅拌时间过短,药粉混合不均匀,外观色泽不均一。注模温度对灌装成型有直接影响,温度过高,灌注后冷却时间较长,雄黄容易沉降,导致溶液分层,造成含量不均匀;温度过低,灌注时容易造成栓剂中空或者残缺,不合格产品增多[8]。通过考察不同制备条件对二黄栓制备工艺的影响,得最佳制备工艺,对二黄栓的工业化生产具有较好的实际应用价值,并为同类栓剂的制备提供参考。
3.4不同前处理方法对砷含量测定的影响 目前砷含量测定方法主要有电感耦合等离子体质谱法(inductively coupled plasma mass spectrometry,ICP-MS)[9-10]、AAS[11-13]、碘量法[14]等。ICP-MS检出限低,精密度好,能同时测定多元素,但当样品中砷含量较高时,容易污染仪器,昂贵的价格也影响了其通用性[9]。AAS专属性强,灵敏度高,选用火焰原子化器测定,操作步骤简单,满足实验需求[13]。滴定分析法灵敏度比AAS低,但因具有准确、快捷、节约等优点使其成为检测分析领域的经典方法,常用于矿物药的含量测定[15]。《中华人民共和国药典》(2020年版)一部采用经典的分析方法碘量法对雄黄中As2S2进行含量测定[6]。本研究采用碘量法和AAS测定样品,结果表明无显著性差异,说明两种方法均适用于本栓中As2S2的含量测定。根据不同检测方法结合特定的前处理方法,使其含量测定更具准确性和科学性,为中成药中矿物药的含量测定提供借鉴。
猜你喜欢
中国西部(2022年2期)2022-05-23 13:28:20
南大法学(2021年6期)2021-04-19 12:27:30
中华养生保健(2020年4期)2020-11-16 01:31:44
活力(2019年15期)2019-09-25 07:22:12
测控技术(2018年6期)2018-11-25 09:50:24
中国测试(2018年9期)2018-05-14 15:33:32
家庭医药·快乐养生(2017年5期)2017-05-18 11:07:56
河北地质(2016年4期)2016-03-20 13:52:08
发明与创新(2015年30期)2015-02-27 10:39:53
腐植酸(2014年6期)2014-04-18 08:57:03
