
线粒体基因突变所致的遗传性线粒体病
2023-11-02 05:59梁少姗曾彩虹
肾脏病与透析肾移植杂志 2023年5期
梁少姗 张 炯 曾彩虹
[作者单位]东部战区总医院 国家肾脏疾病临床医学研究中心(南京,210016)
病例摘要
现病史女性,19岁,因“体检发现尿蛋白阳性伴血尿酸高6年余”于2021-07-26入院。患者诉2015年体检发现尿蛋白1+,血清肌酐(SCr)正常,血尿酸520 μmol/L,予低嘌呤饮食。2015—2021年间多次复查尿蛋白阴性至3+,尿蛋白定量0.517 g/d,尿隐血阴性,SCr 64~66 μmol/L,血尿酸430~525 μmol/L,2020年8月空腹血糖5.66 mmol/L,予中药汤剂(具体不详)治疗10月。2021年7月外院复查尿蛋白定量1.05 g/d,尿隐血阴性,SCr 64.5 μmol/L,血尿酸510.4 μmol/L,CT示髓质海绵肾可能。为进一步诊治入院。患者病程中精神尚可,体力、食欲和睡眠正常,体重无明显变化,大便和排尿正常。
既往史患者足月出生,体重3 100 g。智力发育正常。8岁时因身高发育欠佳至儿童医院间断予以生长激素治疗约6年。2017-07-01起服用 “优甲乐”25 μg/d(使用时间不详)。听力下降多年,既往检查提示神经性耳鸣。否认肝炎、结核、疟疾等传染病史,否认“高血压”等病史,否认手术史、外伤史、输血史,否认药物、食物过敏史,预防接种史不详。
个人史、月经史无特殊,未婚未育。
家族史独生女,父母均体健,父亲身高178 cm,母亲身高150 cm,无不适症状,定期体检正常。
体格检查体温36.3 ℃,脉搏84 次/min,呼吸18次/min,血压106/84 mmHg,身高150 cm,体重50 kg,体表面积1.4 m2,体质量指数(BMI)22.22 kg/m2。神志清,心肺查体未及明显异常,腹软、无压痛,肝脾肋下未触及,双下肢无水肿。
实验室检查
血常规 白细胞计数7.20×109/L,红细胞计数4.19×1012/L,血红蛋白129 g/L,血小板计数329×109/L。
尿液检查 尿蛋白定量0.97 g/d,尿沉渣红细胞7.6/μL,白细胞10.00/μL。尿N-乙酰-β-D-氨基葡萄糖苷酶10.8 U/(g·Cr),尿视黄醇结合蛋白 0.15 mg/L,中性粒细胞明胶酶相关脂质运载蛋白 30 ng/mL,胱抑素C 0.35 mg/L。尿C3 2.00 mg/L,α2-微球蛋白2.00 mg/L。24h尿葡萄糖51.5 mmol/L,尿酸1 830.0 μmol/L,尿钾15.4 mmol/L,尿钠 29.50 mmol/L,尿磷 7.2 mmol/L,尿氯24.0 mmol/L,尿钙0.34 mmol/L,24h尿量1 650 mL。
血生化 血清白蛋白44.30 g/L,球蛋白21.2 g/L,SCr 65 μmol/L,尿素氮5.14 mmol/L,尿酸453 μmol/L,总胆固醇 4.23 mmol/L,三酰甘油2.01 mmol/L,钾4.42 mmol/L,钠 146.0 mmol/L,氯 106.8 mmol/L,钙 2.31 mmol/L,胱抑素C 0.84 mg/L,估算的肾小球滤过率(eGFR)(CKD-EPI) 118 mL/(min·1.73m2)。C反应蛋白<0.5 mg/L。空腹血糖波动于7.7~10.8 mmol/L,餐后2 h血糖波动于8.2~17.4 mmol/L,糖化血红蛋白(HbA1c) 7.3%,胰岛素 7.61 mIU/L,C肽 3.32 ng/mL,胰岛素抗体 2.25 IU/mL。
免疫学检查 抗核抗体<1∶100,抗双链DNA抗体 <1∶10,补体C3 1.2560 g/L,C4 0.2370 g/L。抗磷脂酶A2受体抗体 <2.00 RU/mL。IgG 11.00 g/L,IgA 1.20 g/L,IgM 1.070 g/L,IgE 620.0 IU/mL。甲状腺功能五项未见明显异常。
其他 粪常规+隐血未见明显异常。
辅助检查
肾脏超声 左肾:103 mm×47 mm×51 mm,右肾:100 mm×45 mm×50mm,皮质回声增强,皮髓界限清楚,集合系统正常,左肾内可见多个类圆形无回声区,较大的约8 mm×8 mm,部分囊周伴钙化,界清,左肾内可见数个点状强回声,未见后声影
心脏 超声:大致正常。心电图:窦性心律,预激综合征。
胸部 CT右肺下叶少许纤维条索;左肾髓质多发钙化影,考虑髓质海绵肾可能
其他 眼底:视网膜正常,视乳头边界清,黄斑区正常,动静脉比例正常,动脉硬化程度正常。耳鼻喉专科检查示双耳鼓膜完整、标志清,纯音测听示高频听力下降。
外周血基因检测分析到线粒体MT-TL1基因上存在m.3243A>G突变,突变频率为35374/(35374+29211)(55.8%),为致病性变异,经家系验证分析,其母亲该位点突变频率为4485/(4485+58680)(7.1%)。
肾活检
光镜 24个肾小球中5个球性废弃,2个节段硬化,节段硬化处周围可见细胞增生(图1A)。余肾小球系膜区偶见增宽,毛细血管袢开放好,球门部可见透明滴,囊壁节段增厚。PASM-Masson:阴性。肾小管间质轻度慢性病变(5%),小灶性肾小管萎缩、基膜增厚,散在肾小管上皮细胞刷状缘脱落,管腔内少量蛋白管型,间质散在单个核细胞浸润。动脉未见明确病变。
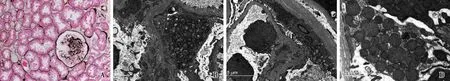
图1 肾小球节段硬化,节段硬化处周围可见细胞增生,肾小管上皮细胞胞质未见明确病变(A:PASM-Masson,×200);肾小球足细胞(B)、壁层上皮细胞(C)、肾小管上皮细胞(D)胞质内异常线粒体(EM)
免疫荧光 IgA trace、IgM trace,呈颗粒状节段分布于系膜区。IgG、C3、C1q、κ轻链、λ轻链阴性。
电镜 2个肾小球,其中1个见节段硬化。2个肾小球足细胞足突融合范围不一致,分别为80%~90%及40%~50%,胞质少量微绒毛化,见大空泡形成。肾小球系膜区略增宽,系膜基质略增多,系膜区未见电子致密物沉积。肾小球毛细血管袢开放好,基膜内皮下和上皮侧未见电子致密物沉积。基膜厚250~410 nm。肾小球部分足细胞(图1B)、壁层上皮细胞(图1C),个别内皮细胞、系膜细胞胞质内见较多线粒体。肾小管上皮细胞(图1D)胞质内部分线粒体体积增大,形态不规则,嵴减少或排列紊乱。
小结 肾活检病理表现为局灶节段性肾小球硬化(FSGS),肾小球多处足细胞、壁层上皮细胞、肾小管上皮细胞胞质内线粒体异常。
最后诊断遗传性线粒体病(m.3243A>G突变):(1)FSGS,CKD 1期;(2)高尿酸血症;(3)身材矮小;(4)线粒体糖尿病;(5)高频听力下降;(6)预激综合征。
讨 论
患者慢性病程,少年期起病,临床表现为多系统损害。肾脏表现为少至中等量尿蛋白,无镜下血尿,SCr正常,高尿酸血症,血压正常,无贫血,肾活检光镜表现为FSGS。肾外表现内分泌异常(身材矮小,糖尿病),耳聋(高频听力下降),心脏受累(预激综合征)。家族中母亲身材矮小。
根据病因可将FSGS分为4类[1]:(1)原发性FSGS,电镜下足细胞足突广泛融合,常急性起病,激素敏感;(2)遗传性FSGS,包括家族性、散发性、综合征型;(3)继发性FSGS,包括病毒感染、药物或继发于肾小球高滤过因素(如肥胖),表现为肾单位正常或减少,肾小球足细胞足突节段融合,非肾病范围的蛋白尿;(4)病因不明的FSGS,肾小球足细胞足突节段融合,非肾病范围的蛋白尿,未找到继发因素。本例患者呈现多系统受累的综合征型临床表现,且家族中母亲同样表现为身材矮小,因此,考虑遗传性FSGS。
本例患者肾脏病理电镜下突出表现为肾小球足细胞、壁层上皮细胞、肾小管上皮细胞胞质内线粒体大小、多少和形态异常。肾脏细胞内线粒体异常可分为遗传性和继发性两类。前者可由线粒体DNA(mtDNA)或核DNA(nDNA)突变引起,后者常见药物(如阿德福韦酯、替诺福韦、异磷酰胺、慢性锂中毒、环孢素A等)、重金属(如镉等)、脓毒血症等。该患者未发现药物、毒物、重金属等接触史。结合患者多系统受累,肾活检病理形态学及家族史特点,基因检测分析到线粒体MT-TL1基因上存在m.3243A>G突变,母系遗传,最终诊断为线粒体基因突变所致的遗传性线粒体病。
遗传性线粒体病通常指的是一类由于遗传突变导致线粒体功能障碍引发的以多系统细胞呼吸链及能量代谢异常为特点的疾病。任何年龄均可发病,大多累及多个器官,高耗能器官优先受累,如骨骼肌、心脏、神经系统、肾脏。此外,还累及内分泌系统、眼、消化道、肝脏等,不同的位点突变可呈特定综合征症候群,如本例表现为母系遗传性耳聋和糖尿病(MIDD),常可延迟诊断。
线粒体病肾脏损害,儿童期发病者常为散发,通常年龄小,Fanconi综合征最常见,其他可表现为激素抵抗的肾病综合征、Batter样综合征及肾小管酸中毒等,出现全身器官受累时常已进展为肾功能不全,基因突变包括mtDNA及nDNA,合成辅酶Q10(CoQ10)相关nDNA基因突变多见。成人期发病者母系遗传占优势,发病年龄不定,多数患者以肾脏损害为首发表现,听力下降尤为多见,但严重多器官受累少见,线粒体MT-TL1基因中的m.3243A>G突变为热点突变。一项纳入81例线粒体肾病的研究发现[2],m.3243A>G突变占78%,该突变的患者约半数有线粒体病家族史,60%患者有糖尿病家族史,常表现为少量至中等量蛋白尿、肾功能不全,少数为Fanconi综合征,亦见高尿酸血症[3],伴发的肾外表现最常见为听力障碍、糖尿病,其次为发育障碍、心肌病、癫痫。此外,线粒体肾病的肾脏影像学检查可表现为肾囊性变,应注意髓质海绵肾相鉴别。
遗传性线粒体病肾脏病理半数表现为FSGS,其次为糖尿病肾病、肾硬化、肾小管间质性肾炎、病变轻微或者肾囊肿,仅22%的患者在光镜下可见胞浆肿胀、颗粒变性的上皮细胞。电镜下57%患者见异常线粒体,约1/3位于足细胞、肾小管上皮细胞,少数位于内皮细胞、系膜细胞、动脉平滑肌细胞、壁层上皮细胞[2,4]。此外,线粒体病肾脏损害在肾活检病理上可合并其他肾脏疾病,如IgA肾病[5],膜性肾病等[6]。由于组织学改变不具有特异性,有时可合并其他肾脏疾病,超微结构上不一定能观察到异常线粒体等,使得通过肾脏病理协助诊断线粒体病具有一定的挑战性。
线粒体疾病基因检测采用高通量测序,识别mtDNA的点突变、片段缺失或插入、线粒体拷贝数变异,以及编码线粒体呼吸链的nDNA突变。mtDNA具有突变异质性,即同一细胞或组织中同时存在突变型和野生型mtDNA,当突变的mtDNA比例在相应器官中达到一定阈值时,才出现临床症状,因此,需明确样本的mtDNA突变比例。本中心既往研究发现,8例mtDNA突变相关线粒体肾病患者共有3个位点突变表现出异质性,其中m.3243A>G突变的病例外周血的mtDNA突变频率较低(37%~61%),肾组织和尿液的突变频率均>70%(76%~90%)。值得注意的是,对于一些mtDNA突变,致病突变频率可能动态下降,特别是在快速分裂组织(如血液前体细胞分裂产生的外周血细胞[6]),老年患者可能难以通过外周血检测出m.3243A>G突变。此时,可通过遗传变异性较小的组织细胞(如骨骼肌细胞、尿液上皮细胞等)检测此特定突变[7]。本例患者仅检测了外周血的线粒体,突变频率为55.8%,其母亲突变频率为7.1%,均未行肾组织或尿液的基因检测。
线粒体病的临床表现具有多样性,人们提出了数个诊断标准,综合了临床表型、代谢和影像学、病理和分子特征,对其进行评分后进行诊断分级[8-10]。基因诊断的临床普及为线粒体病的临床诊断提供关键依据。对Morava诊断标准进行再验证发现,基因确诊的线粒体病患者诊断分级分别为明确(51%)或可能(33%)[11]。本例患者的在上述三个诊断标准分级均为可能,最终经基因检测证实。
该病以器官支持治疗为主,补充辅酶Q10及其他参与呼吸链的抗氧化剂或辅助因子也可能提供一定的临床获益[12],适当避免可能加剧疾病进展的药物。随着时间的推移,mtDNA突变的线粒体病患者可能发展出更多器官的功能失调(终末分化的组织细胞可能突变频率增加[13]),因此仍需定期进行主要器官受累的筛查[14]。研究发现m.3243A>G突变患者,半数进入终末期肾病,25%死亡(中位随访时间分别为11年及12年)。终末期肾病患者可进行透析,大多数接受肾移植的病例表现良好[15],肾移植前应多学科讨论,如心功能评估、麻醉方案等,母系供肾仍存在争议[16]。女性妊娠产科并发症风险增加,有低出生体重儿童、死胎风险[17]。卵子胞浆置换技术辅助生育可潜在防止mtDNA突变从母亲传递到后代。
小结:本文报道了1例母系遗传的线粒体MT-TL1基因m.3243A>G突变所致的线粒体病。FSGS是具有异质性的形态学改变,当临床表现为多系统受累时,应注意排查遗传性疾病,超微结构观察到线粒体异常给疾病诊断提供重要线索,结合家族史、基因检测等进一步明确诊断。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
昆明医科大学学报(2022年4期)2022-05-23
基层中医药(2021年8期)2021-11-02
中外医疗(2016年15期)2016-12-01
中外医疗(2015年11期)2016-01-04
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
西南军医(2015年6期)2015-01-23
西南军医(2015年1期)2015-01-22
